Cone dystrophy
| Cone dystrophy | |
|---|---|
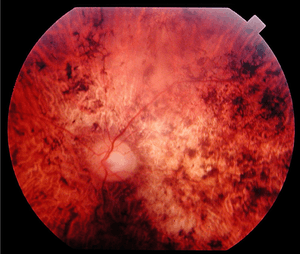 | |
| Fundus of a 34-year-old patient with cone rod dystrophy due to Spinocerebellar Ataxia Type 7 (SCA7). Note that the macular area, and also the mid periphery, are atrophic. | |
| Classification and external resources | |
| Specialty | ophthalmology |
| ICD-10 | H35.5 |
| OMIM | 300085 |
A cone dystrophy is an inherited ocular disorder characterized by the loss of cone cells, the photoreceptors responsible for both central and color vision.
The most common symptoms of cone dystrophy are vision loss (age of onset ranging from the late teens to the sixties), sensitivity to bright lights, and poor color vision. Therefore, patients see better at dusk. Visual acuity usually deteriorates gradually, but it can deteriorate rapidly to 20/200; later, in more severe cases, it drops to "counting fingers" vision. Color vision testing using color test plates (HRR series) reveals many errors on both red-green and blue-yellow plates.
The pathogenesis of cone dystrophy has yet to be elucidated. It appears that the dystrophy is primary, since subjective and objective abnormalities of cone function are found before ophthalmoscopic changes can be seen. However, the retinal pigment epithelium (RPE) rapidly becomes involved, leading to a retinal dystrophy primarily involving the macula. The histological examination of the eyes of one such patient showed that the outer nuclear layer of cones and rods had disappeared completely, whereas the RPE showed pronounced pigment changes. There was also atrophy of the temporal disc.
The fundus exam via ophthalmoscopy is essentially normal early on in cone dystrophy, and definite macular changes usually occur well after visual loss. Fluorescein angiography (FA) is a useful adjunct in the workup of someone suspected to have cone dystrophy, as it may detect early changes in the retina that are too subtle to be seen by ophthalmoscope. For example, FA may reveal areas of hyperfluorescence, indicating that the RPE has lost some of its integrity, allowing the underlying fluorescence from the choroid to be more visible. These early changes are usually not detected during the ophthalmoscopic exam.
The most common type of macular lesion seen during ophthalmoscopic examination has a bull’s-eye appearance and consists of a doughnut-like zone of atrophic pigment epithelium surrounding a central darker area. In another, less frequent form of cone dystrophy there is rather diffuse atrophy of the posterior pole with spotty pigment clumping in the macular area. Rarely, atrophy of the choriocapillaris and larger choroidal vessels is seen in patients at an early stage. The inclusion of fluorescein angiography in the workup of these patients is important since it can help detect many of these characteristic ophthalmoscopic features. In addition to the retinal findings, temporal pallor of the optic disc is commonly observed.
As expected, visual field testing in cone dystrophy usually reveals a central scotoma. In cases with the typical bull’s-eye appearance, there is often relative central sparing.
Because of the wide spectrum of fundus changes and the difficulty in making the diagnosis in the early stages, electroretinography (ERG) remains the best test for making the diagnosis. Abnormal cone function on the ERG is indicated by a reduced single-flash and flicker response when the test is carried out in a well-lit room (photopic ERG). The relative sparing of rod function in cone dystrophy is evidenced by a normal scotopic ERG, i.e. when the test is carried out in the dark. In more severe or longer standing cases, the dystrophy involves a greater proportion of rods with resultant subnormal scotopic records. Since cone dystrophy is hereditary and can be asymptomatic early on in the disease process, ERG is an invaluable tool in the early diagnosis of patients with positive family histories.
Cone dystrophy in general usually occurs sporadically. Hereditary forms are usually autosomal dominant, and instances of autosomal recessive and X-linked inheritance also occur.
In the differential diagnosis, other macular dystrophies as well as the hereditary optic atrophies must be considered. Fluorescent angiography, ERG, and color vision tests are important tools to help facilitate diagnosis in early stages.
Genetics
At least one type of autosomal dominant cone-rod dystrophy is caused by mutations in the guanylate cyclase 2D gene (GUCY2D) on chromosome 17.
Treatment
Though there is no treatment for Cone dystrophy, certain supplements may help in delaying the progression of the disease.
The beta-carotenoids, lutein and zeaxanthin, have been evidenced to reduce the risk of developing age related macular degeneration (AMD),[1] and may therefore provide similar benefits to Cone dystrophy sufferers.
Consuming omega-3 fatty acids (docosahexaenoic acid and eicosapentaenoic acid) has been correlated with a reduced progression of early AMD, and in conjunction with low glycemic index foods, with reduced progression of advanced AMD,[2] and may therefore delay the progression of cone dystrophy.
Notes
- ↑ Carpentier S, Knaus M, Suh M (2009). "Associations between lutein, zeaxanthin, and age-related macular degeneration: An overview". Critical reviews in Food Science and Nutrition. 49 (4): 313–326. doi:10.1080/10408390802066979. PMID 19234943.
Abstract doesn't include conclusion
- ↑ Chiu CJ, Klein R, Milton RC, Gensler G, Taylor A (June 2009). "Does eating particular diets alter the risk of age-related macular degeneration in users of the Age-Related Eye Disease Study supplements?". Br J Ophthalmol. 93 (9): 1241–6. doi:10.1136/bjo.2008.143412. PMC 3033729
 . PMID 19508997.
. PMID 19508997. Conclusions: The findings show an association of consuming a diet rich in DHA with a lower progression of early AMD. In addition to the AREDS supplement, a lower dGI with higher intakes of DHA and EPA was associated with a reduced progression to advanced AMD.
References
- Stephen J. Ryan et al., Retina, 3rd ed. (C.V. Mosby, 2001) ISBN 0-323-00804-6
- Stephen J. Ryan et al., Retina, 4th ed. (C.V. Mosby, 2005) ISBN 0-323-02598-6
- Carpentier S, Knaus M, Suh M (2009). "Associations between lutein, zeaxanthin, and age-related macular degeneration: An overview". Critical reviews in Food Science and Nutrition 49 (4): 313–326. doi:10.1080/10408390802066979. PMID 19234943. "Abstract doesn't include conclusion"
- Chiu CJ, Klein R, Milton RC, Gensler G, Taylor A (June 2009). "Does eating particular diets alter the risk of age-related macular degeneration in users of the Age-Related Eye Disease Study supplements?". Br J Ophthalmol 93 (9): 1241–6. doi:10.1136/bjo.2008.143412. PMC 3033729. PMID 19508997. "Conclusions: The findings show an association of consuming a diet rich in DHA with a lower progression of early AMD. In addition to the AREDS supplement, a lower dGI with higher intakes of DHA and EPA was associated with a reduced progression to advanced AMD."