Congenital adrenal hyperplasia due to 11β-hydroxylase deficiency
| Congenital adrenal hyperplasia due to 11β-hydroxylase deficiency | |
|---|---|
| Classification and external resources | |
| OMIM | 202010 |
| eMedicine | article/117012 |

11β-Hydroxylase deficient congenital adrenal hyperplasia (11β-OH CAH) is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a defect in the gene encoding the enzyme steroid 11β-hydroxylase which mediates the final step of cortisol synthesis in the adrenal. 11β-OH CAH results in hypertension due to excessive mineralocorticoid effects. It also causes excessive androgen production both before and after birth and can virilize a genetically female fetus or a child of either sex.
Classification
Congenital adrenal hyperplasia (CAH) refers to any of several autosomal recessive diseases resulting from defects in steps of the synthesis of cortisol from cholesterol by the adrenal glands. All of the forms of CAH involve excessive or defective production of sex steroids and can prevent or impair development of primary or secondary sex characteristics in affected infants, children, and adults. Many also involve excessive or defective production of mineralocorticoids, which can cause hypertension or salt wasting, respectively.
The most common type of CAH is due to deficiency of 21-hydroxylase. 11β-Hydroxylase deficient congenital adrenal hyperplasia is one of the less common types of CAH due to deficiencies of other proteins and enzymes involved in cortisol synthesis. Other uncommon types are described in individual articles (links below).
11β-OH CAH resembles 21-hydroxylase deficient CAH in its androgenic manifestations: partial virilization and ambiguous genitalia of genetically female infants, childhood virilization of both sexes, and rarer cases of virilization or infertility of adolescent and adult women. The mineralocorticoid effect differs: hypertension is usually the clinical clue that a patient has 11- rather than 21-hydroxylase CAH. Diagnosis of 11β-OH CAH is usually confirmed by demonstration of marked elevations of 11-deoxycortisol and 11-deoxycorticosterone (DOC), the substrates of 11β-hydroxylase. Management is similar to that of 21-hydroxylase deficient CAH except that mineralocorticoids need not be replaced.
Characteristics
Mineralocorticoid effects
Mineralocorticoid manifestations of severe 11β-hydroxylase deficient CAH can be biphasic, changing from deficiency (salt-wasting) in early infancy to excess (hypertension) in childhood and adult life.
Salt-wasting in early infancy does not occur in most cases of 11β-OH CAH but can occur because of impaired production of aldosterone aggravated by inefficiency of salt conservation in early infancy. When it occurs it resembles the salt-wasting of severe 21-hydroxylase deficient CAH: poor weight gain and vomiting in the first weeks of life progress and culminate in life-threatening dehydration, hyponatremia, hyperkalemia, and metabolic acidosis in the first month.
Despite the inefficient production of aldosterone, the more characteristic mineralocorticoid effect of 11β-OH CAH is hypertension. Progressive adrenal hyperplasia due to persistent elevation of ACTH results in extreme overproduction of 11-deoxycorticosterone (DOC) by mid-childhood. DOC is a weak mineralocorticoid, but usually reaches high enough levels in this disease to cause effects of mineralocorticoid excess: salt retention, volume expansion, and hypertension.
Sex steroid effects
Because 11β-hydroxylase activity is not necessary in the production of sex steroids (androgens and estrogens), the hyperplastic adrenal cortex produces excessive amounts of DHEA, androstenedione, and especially testosterone.
These androgens produce effects that are similar to those of 21-hydroxylase deficient CAH. In the severe forms, XX (genetically female) fetuses can be markedly virilized, with ambiguous genitalia that look more male than female, though internal female organs, including ovaries and uterus develop normally.
XY fetuses (genetic males) typically show no abnormal features related to androgen excess. A megalopenis (>22 cm/8.7in) is usually present in male patients.
In milder mutations, androgen effects in both sexes appear in mid-childhood as early pubic hair, overgrowth, and accelerated bone age. Although "nonclassic" forms causing hirsutism and menstrual irregularities and appropriate steroid elevations have been reported, most have not had verifiable mutations and mild 11β-hydroxylase deficient CAH is currently considered a very rare cause of hirsutism and infertility.
All of the issues related to virilization, neonatal assignment, advantages and disadvantages of genital surgery, childhood and adult virilization, gender identity and sexual orientation are similar to those of 21-hydroxylase CAH and elaborated in more detail in Congenital adrenal hyperplasia.
Pathophysiology
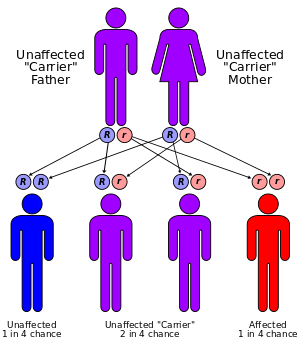
The enzyme which mediates 11β-hydroxylase activity is now known as P450c11β since it is one of the cytochrome P450 oxidase enzymes located in the inner mitochondrial membrane of cells of the adrenal cortex. It is coded by a gene at 8q21-22. Like the other forms of CAH, a number of different defective alleles for the gene have been identified, producing varying degrees of impaired 11β-hydroxylase activity. Also like the other forms of CAH, 11β-OH CAH is inherited as an autosomal recessive disease.
11β-Hydroxylase mediates the final step of the glucocorticoid pathway, producing cortisol from 11-deoxycortisol. It also catalyzes the conversion of 11-deoxycorticosterone (DOC) to corticosterone in the mineralocorticoid pathway.
Management
As with other forms of CAH, the primary therapy of 11β-hydroxylase deficient CAH is lifelong glucocorticoid replacement in sufficient doses to prevent adrenal insufficiency and suppress excess mineralocorticoid and androgen production.
Salt-wasting in infancy responds to intravenous saline, dextrose, and high dose hydrocortisone, but prolonged fludrocortisone replacement is usually not necessary. The hypertension is ameliorated by glucocorticoid suppression of DOC.
Long term glucocorticoid replacement issues are similar to those of 21-hydroxylase CAH, and involve careful balance between doses sufficient to suppress androgens while avoiding suppression of growth. Because the enzyme defect does not affect sex steroid synthesis, gonadal function at puberty and long-term fertility should be normal if adrenal androgen production is controlled. See congenital adrenal hyperplasia for a more detailed discussion of androgen suppression and fertility potential in adolescent and adult women.
See also
- Inborn errors of steroid metabolism
- Congenital adrenal hyperplasia
- Adrenal insufficiency
- Disorders of sexual development
- Intersexuality, pseudohermaphroditism, and ambiguous genitalia
- Steroid 11β-hydroxylase