Death-inducing signaling complex
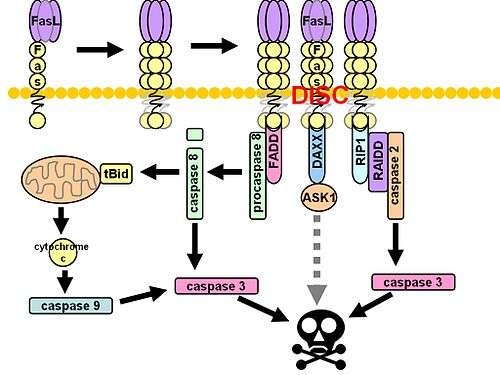
The death-inducing signaling complex or DISC is a multi-protein complex formed by members of the "death receptor" family of apoptosis-inducing cellular receptors.[1] A typical example is FasR, which forms the DISC upon trimerization as a result of its ligand (FasL) binding. The DISC is composed of the death receptor, FADD, and caspase 8. It transduces a downstream signal cascade resulting in apoptosis.
The Fas ligands, or cytotoxicity-dependent APO-1-associated proteins, physically associate with APO-1 (also known as the Fas receptor, or CD95), a tumor necrosis factor containing a functional death domain. This association leads to the formation of the DISC, thereby inducing apoptosis.[2] The entire process is initiated when the cell registers the presence of CD95L, the cognate ligand for APO-1.[3] Upon binding, the CAP proteins and procaspase-8 (composed of FLICE, MACH, and Mch5) bind to CD95 through death domain and death effector domain interactions. Procaspase-8 activation is thought to occur through a dimerization process with other procaspase-8 molecules, known as an induced proximity model.
The CAP proteins associate only with the oligomerized version of APO-1 when forming the complex. The CAP1 are CAP2 proteins are also known as FADD/MORT1, an adaptor molecule with a death domain. CAP4 is also called FLICE, a cysteine protease with two death effector domains.[4] CAP3 is the prodomain of FLICE generated during proteolytic activation.[5] Once the DISC assembles, it allows APO-1 signaling to occur, which triggers cell death. In order to do this, downstream targets such as FLICE must be activated. In its inactive state, FLICE's two death domains are thought to bind together and prevent its activation. Once APO-1 aggregates within the cytosol, it recruits FADD, CAP3, and FLICE to the receptor, where FLICE is modified into several active subunits, which have the ability to cleave a variety of substrates. This proteolytic activity then results in a cascade of caspase activation, and ultimately cell death. This apoptotic activity is critical for tissue homeostasis and immune function.
APO-1-mediated apoptosis can be inhibited by a variety of factors, including the viral caspase inhibitors CrmA and p35, as well as viral FLICE-inhibitory proteins known as v-FLIPs. When in the presence of APO-1, v-FLIPs preferentially bind and prevent procaspase-8 from being recruited; as such, apoptosis is stalled. Interestingly, humans have a homolog for v-FLIP known as c-FLIP, which occurs in two endogenous forms (c-FLIPL (long) and c-FLIPS (short)). These are similar in structure to procaspase-8, but lack the amino acids necessary for caspase-8 catalytic activity. It is thought that c-FLIP may be involved in modulating the immune system, as c-FLIPS is upregulated upon stimulation of the T cell receptor. Furthermore, as high expression of FLIP is known to promote tumor growth, these inhibitor molecules play a role in cancer proliferation.
The DISC has been implicated as a possible drug development target for various cancers, including leukemia, glioma, and colon cancer. In glioma cells, the effects of TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) have been shown to induce DISC-mediated apoptosis. Specifically, TRAIL works by activating two death receptors, DR4 and DR5; these bind to FADD, which then interacts with caspase-8 to assemble the DISC. Tumor cells show varying sensitivity to TRAIL modulated apoptosis, depending on the presence of the antiapoptotic FLIP proteins.[6] Additionally, studies in leukemia have indicated that the histone deacetylase inhibitor LAQ824 increases apoptosis by decreasing the expression levels of the c-FLIPs.[7] As such, these inhibitors are promising targets for anti-cancer therapy.
References
- ↑ Kischkel, FC; Hellbardt, S; Behrmann, I; Germer, M; Pawlita, M; Krammer, PH; Peter, ME (Nov 15, 1995). "Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor.". The EMBO Journal. 14 (22): 5579–88. PMC 394672
 . PMID 8521815.
. PMID 8521815. - ↑ Kischkel, FC; Hellbardt, S; Behrmann, I; Germer, M; Pawlita, M; Krammer, PH; Peter, ME (Nov 15, 1995). "Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor.". The EMBO Journal. 14 (22): 5579–88. PMC 394672. PMID 8521815.
- ↑ Krueger, A; Schmitz, J; Baumann, S; Krammer, P; Kirchoff, S (Jun 8, 2001). "Cellular FLICE-inhibitory Protein Splice variants Inhibit Different Steps of Caspase-8 Activation at the CD95 Death-inducing Signaling Complex.". Journal of Biological Chemistry. 276: 20633–40. doi:10.1074/jbc.M101780200. PMID 11279218.
- ↑ Medema, JP; Scaffidi, C; Kischkel, F; Shevencho, A; Mann, M; Krammer, P; Peter, ME (May 15, 1997). "FLICE is activated by association with the CD95 death-inducing signaling complex (DISC).". The EMBO Journal. 16 (10): 2794–2804. doi:10.1093/emboj/16.10.2794. PMC 1169888. PMID 9184224.
- ↑ Muzio, M; Chinnaiyan, A; Kischkel, F; O'Rourke, K; Shevchenko, A; Ni, J; Dixit, V (Jun 14, 1996). "FLICE, A Novel FADD-Homologous ICE/CED-3-like Protease, is Recruited to the CD95 (Fas/APO-1) Death-Inducing Signaling Complex.". Cell. 85 (6): 817–27. doi:10.1016/s0092-8674(00)81266-0. PMID 8681377.
- ↑ Xiao, C; Yang, B; Asadi, N; Beguinot, F; Hao, C (Apr 25, 2002). "Tumor Necrosis Factor-related Apoptosis-inducing Ligand-induced Death-inducing Signaling Complex and its Modulation by c-FLIP and PED/PEA-15 in Glioma Cells.". The Journal of Biological Chemistry. 277: 25020–25. doi:10.1074/jbc.M202946200. PMID 11976344.
- ↑ Cuo, S; Sigua, C; Tao, J; Bali, P; George, P; Li, Y; Bhalla, K (Apr 1, 2004). "Cotreatment with Histone Deacetylase Inhibitor LAQ824 Enhances Apo-2L/Tumor Necrosis Factor-Related Apoptosis Inducing Ligand-Induced Death Inducing Signaling Complex Activity and Apoptosis of Human Acute Leukemia Cells.". Cancer Research. 64: 2580–9. PMID 15059915.
External links
- DISC (Death Inducing Signaling Complex) at the US National Library of Medicine Medical Subject Headings (MeSH)