Lattice corneal dystrophy
| Lattice corneal dystrophy type | |
|---|---|
 | |
| A network of thick linear corneal opacities in patient with a variant of LCD1 (LCD type III) due to a homozygous p. Leu527Arg mutation in the TGFBI gene | |
| Classification and external resources | |
| Specialty | ophthalmology |
| ICD-10 | H18.5 |
| ICD-9-CM | 371.54 |
| OMIM | 122200 |
| eMedicine | article/1193793 |
Lattice corneal dystrophy type, also known as Biber-Haab-Dimmer dystrophy, is a rare form of corneal dystrophy. It has no systemic manifestations, unlike the other type of the dystrophy, Lattice corneal dystrophy type II. Lattice corneal dystrophy was first described by Swiss ophthalmologist Hugo Biber in 1890.[1]
Lattice dystrophy gets its name from an accumulation of amyloid deposits, or abnormal protein fibers, throughout the middle and anterior stroma.
Genetics

Lattice corneal dystrophy has two types:[2]
- type I: with no systemic association. It is caused by mutations in TGFBI gene encoding keratoepithelin, which maps to chromosome 5q.
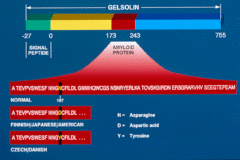
- type II or Finnish type amyloidosis: associated with manifestations of systemic amyloidosis due to accumulation of gelsolin.[3] Associated conditions may include cutis laxa[4] and ataxia.[5]
- type III is also described which has an onset at age 70 to 90 years and is not associated with systemic amyloidosis.[2]
Clinical presentation
Filamentous opacities appear in the cornea with intertwining delicate branching processes. During an eye examination, the doctor sees these deposits in the stroma as clear, comma-shaped overlapping dots and branching filaments, creating a lattice effect. Over time, the lattice lines will grow opaque and involve more of the stroma. They will also gradually converge, giving the cornea a cloudiness that may also reduce vision. The disease is bilateral, usually noted before the end of the first decade of life. Although lattice dystrophy can occur at any time in life, the condition usually arises in children between the ages of two and seven.
In some people, these abnormal protein fibers can accumulate under the cornea's outer layer—the epithelium. This can cause erosion of the epithelium. This condition is known as recurrent epithelial erosion. These erosions alter the cornea's normal curvature, resulting in temporary vision problems, and expose the nerves that line the cornea, causing severe pain. Even the involuntary act of blinking can be painful.
In systemic cases, kidney failure, heart failure and neuropathy such as facial nerve palsy, laxity of the skin may be noted.[6]
Treatment
In case of corneal erosion, a doctor may prescribe eye drops and ointments to reduce the friction on the eroded cornea. In some cases, an eye patch may be used to immobilize the eyelids. With effective care, these erosions usually heal within three to seven days, although occasional sensations of pain may occur for the next six-to-eight weeks. As patients with LCD suffer with dry eyes as a result of erosion, a new technique involving the insertion of punctal plugs (both upper and lower) can reduce the amount of drops used a day, aiding ocular stability.
By about age 40, some people with lattice dystrophy will have scarring under the epithelium, resulting in a haze on the cornea that can greatly obscure vision. In this case, a corneal transplantation may be needed. There have been many cases in which teenage patients have had the procedure, which accounts for the change in severity of the condition from person to person.
Although people with lattice dystrophy have an excellent chance for a successful corneal transplantation, the disease may also arise in the donor cornea in as little as three years. In one study, about half of the transplant patients with lattice dystrophy had a recurrence of the disease between two and 26 years after the operation. Of these, 15 percent required a second corneal transplant. Early lattice and recurrent lattice arising in the donor cornea responds well to treatment with the excimer laser.
Phototherapeutic keratectomy (PTK) using [Excimer laser] can restore and preserve useful visual function for a significant period of time in patients with anterior corneal dystrophies.[2]
- References
- Maury CPJ, Alli K, Baumann M ( January 1990) Finnish hereditary amyloidosis - amino acid sequence homology between the amyloid fibril protein and human plasma gelsolin. FEBS Letters vol 260,pp 85–87 (1990)
- Maury CPJ, Kere J, Tolvanen R et al. ( Dec 1990) Finnish hereditary amyloidosis is caused by a single nucleotide substitution in the gelsolin gene. FEBS Letters vol 276, pp. 75–77 (1990)
[7]; column-width: refs [7]; list-style-type: decimal;">
- ↑ H. Biber: Über einige seltenere Hornhauterkrankungen. Zürich, 1890. Inaugural Dissertation, Zurich, 1890. Cited by O. Haab: Die gittrige Keratitis.
- 1 2 3 Online Mendelian Inheritance in Man (OMIM) 122200
- ↑ de la Chapelle A, Kere J, Sack GH, Tolvanen R, Maury CP (July 1992). "Familial amyloidosis, Finnish type: G654----a mutation of the gelsolin gene in Finnish families and an unrelated American family". Genomics. 13 (3): 898–901. doi:10.1016/0888-7543(92)90182-R. PMID 1322359.
- ↑ Kiuru-Enari S, Keski-Oja J, Haltia M (February 2005). "Cutis laxa in hereditary gelsolin amyloidosis". Br. J. Dermatol. 152 (2): 250–7. doi:10.1111/j.1365-2133.2004.06276.x. PMID 15727635.
- ↑ Tanskanen M, Paetau A, Salonen O, et al. (March 2007). "Severe ataxia with neuropathy in hereditary gelsolin amyloidosis: a case report". Amyloid. 14 (1): 89–95. doi:10.1080/13506120601116393. PMID 17453628.
- ↑ Online Mendelian Inheritance in Man (OMIM) 105120
- 1 2 Emmett T. Cunningham; Paul Riordan-Eva. Vaughan & Asbury's general ophthalmology. (18th ed.). McGraw-Hill Medical. ISBN 978-0071634205.