Trifluridine/tipiracil
 | |
| Combination of | |
|---|---|
| Trifluridine | Cytotoxin |
| Tipiracil | Thymidine phosphorylase inhibitor |
| Clinical data | |
| Trade names | Lonsurf |
| AHFS/Drugs.com | Lonsurf |
| Pregnancy category |
|
| Routes of administration | By mouth (tablets) |
| ATC code | L01BC59 (WHO) |
| Legal status | |
| Legal status |
|
| Identifiers | |
| Synonyms | TAS-102 |
| CAS Number | 733030-01-8 |
| KEGG | D10526 |
Trifluridine/tipiracil (trade name Lonsurf) is a combination drug for the treatment of metastatic colorectal cancer. It is a combination of two active pharmaceutical ingredients: trifluridine, a nucleoside analog, and tipiracil, a thymidine phosphorylase inhibitor. Tipiracil prevents rapid metabolism of trifluridine, increasing the bioavailability of trifluridine.
Medical uses
The drug is approved for use as a third- or fourth-line treatment in metastatic colorectal cancer patients which have already received both conventional chemotherapy and biologic therapy.[1][2]
In the clinical study in 800 patients on which the approval was based, trifluridine/tipiracil plus best supportive care significantly reduced tumour progression and deaths as compared to placebo plus best supportive care (88.4% versus 94.4% over the course of the study; progression-free survival was 2.0 versus 1.7 months). Overall survival was also improved (7.1 versus 5.3 months).[3][4]
Adverse effects
The most common adverse effects in studies (in more than 10% of patients) were neutropenia, leukopenia, anaemia, thrombopenia, reduced appetite, diarrhoea, nausea, vomiting, and fatigue.[4]
Interactions
Only in vitro interaction studies are available. In these, trifluridine used the concentrative nucleoside transporter 1 (CNT1) and equilibrative nucleoside transporters 1 (ENT1) and 2 (ENT2), and tipiracil was transported by the solute carrier proteins SLC22A2 and SLC47A1. Drugs that interact with these transporters could influence blood plasma concentrations of trifluridine and tipiracil. Trifluridine, being a thymidine phosphorylase inhibitor, could also interact with substrates of this enzyme such as zidovudine.[4]
Pharmacology
Mechanism of action
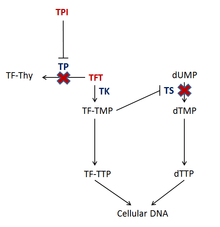
The drug consists of the cytotoxin trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil.[5] Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Trifluridine (TFT) is incorporated into DNA by phosphorylation by thymidylate kinase (TK) to TF-TMP;[6] TF-TMP then covalently binds to tyrosine 146 of the active site of thymidylate synthase (TS) inhibiting the enzyme's activity.[6] TS is vital to the synthesis of DNA because it is an enzyme involved in the synthesis of the deoxynucleotide, thymidine triphosphate (dTTP).[6] Inhibition of TS depletes the cell of dTTP and causes accumulation of deoxyuridine monophosphate (dUMP), which increases the likelihood that uracil gets misincorporated into the DNA.[6] Also, subsequent phosphorylations of TF-TMP cause an increased level of TF-TTP within the cell, which results in it being incorporated into DNA.[6] Even though the exact mechanism of how TFT causes DNA damage is not completely understood, it is hypothesized that the incorporation TF-TTP in DNA leads to DNA strand break formation.[6]
Tipiracil prevents the degradation of trifluridine via thymidine phosphorylase (TP) when taken orally and also has antiangiogenic properties.[6][7][8][9] Not only has thymidine phosphorylase been shown to be identical to platelet-derived endothelial cell growth factor (PD-ECGF), which is an endogenous factor involved in the formation of new vasculature, but also the products of the enzyme may contribute to the stimulation of endothelial cell chemotaxis.[9]
Pharmacokinetics
See Trifluridine#Pharmacokinetics (oral) and Tipiracil#Pharmacokinetics.
History
Since the synthesis of 5-fluorouracil (5-FU) in 1957,[10] fluoropyrimidines have been a useful tool in the treatment of many types of cancer.[11] Due to the drawbacks of 5-FU therapy, such as having to be administered over long periods of time via intravenous infusion and the development of resistance in tumors, more convenient and efficacious fluoropyrimidine therapy has been desired.[11] The fluoropyrimidine component of this drug, trifluridine, was first synthesized in 1964 by Heidelberger et al.[11] By the late 1960s, Phase I and Phase II clinical trials of intravenous trifluridine alone initially proved to be disappointing.[11] Its pharmacokinetic profile during these clinical trials showed that the drug exhibited a very short half-life while in serum (12 minutes post-injection).[11] In response to its pharmacokinetic properties, adjustments in the dosing regimen demonstrated significant therapeutic benefits in patients with breast cancer and colon cancer;[11] with this new regimen, doses would be given every three hours to total a daily amount of 2.5 mg/kg/day for 8 to 13 days, and as a result, eight out of 23 breast cancer patients were reported to have a therapeutic response while one in six patients with colon cancer showed a near complete response to therapy.[11] Success of trifluridine as an effective anti-cancer agent was short lived, however, due to rapid tumor recurrence upon regression of therapy.[11] Trifluridine therapy in oncology was thus halted.[11]
Researchers later found out that trifluridine, when taken orally, was broken down into the inactive metabolites 5-trifluoromethyluracil and 5-trifluoromethyl-2,4(1H,3,H)-pyrimidinedione (FTY) during its extensive first pass metabolism in the liver via the enzyme thymidine phosphorylase.[8][11] It was then hypothesized that orally administered FTD concentrations could be increased and maintained if the drug was given with a thymidine phosphorylase inhibitor.[11]
Trifluridine/tipiracil was approved by the U.S. Food and Drug Administration on 22 September 2015,[1][2] and by the European Medicines Agency on 25 April 2016.[12]
External links
References
- 1 2 "FDA approves new oral medication to treat patients with advanced colorectal cancer" (Press release). Silver Spring, MD. U.S. Food and Drug Administration. 2015-09-22. Retrieved 2015-09-23.
- 1 2 "Taiho Oncology's TAS-102 Meets Primary Endpoint of Improving Overall Survival in Global Phase III RECOURSE Trial in Refractory Metastatic Colorectal Cancer". Taiho Pharma. 12 May 2014. Retrieved 2 December 2014.
- ↑ Burness, C. B.; Duggan, S. T. (2016). "Trifluridine/Tipiracil: A Review in Metastatic Colorectal Cancer". Drugs. 76 (14): 1393–402. doi:10.1007/s40265-016-0633-9. PMID 27568360.
- 1 2 3 Haberfeld, H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag.
- ↑ "A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA". Sep 2004. PMID 15289858.
- 1 2 3 4 5 6 7 Temmink, Olaf (June 2007). "Therapeutic potential of the dual‐targeted TAS‐102 formulation in the treatment of gastrointestinal malignancies". Cancer science. 98 (6): 779–789. doi:10.1111/j.1349-7006.2007.00477.x. PMID 17441963.
- ↑ "New Drug for Colorectal Cancer Shows Promise in Phase II Trial". 28 Aug 2012.
- 1 2 Peters, Godefridus (December 2012). "TAS-102: more than an antimetabolite". The Lancet Oncology. 13 (12): e518–e519. doi:10.1016/s1470-2045(12)70426-6. PMID 23182191.
- 1 2 Matsushita, Shigeto (15 April 1999). "The Effect of a Thymidine Phosphorylase Inhibitor on Angiogenesis and Apoptosis in Tumors". Cancer Research. 59 (8): 1911–1916. PMID 10213500.
- ↑ Hoff, P. M. (1 August 2001). "The Evolution of Fluoropyrimidine Therapy: From Intravenous to Oral". The Oncologist. 6 (90004): 3–11. doi:10.1634/theoncologist.6-suppl_4-3.
- 1 2 3 4 5 6 7 8 9 10 11 Hong, David (15 September 2006). "Phase I Study to Determine the Safety and Pharmacokinetics of Oral Administration of TAS-102 in Patients With Solid Tumors". Cancer. 107 (6): 1383–1390. doi:10.1002/cncr.22125. PMID 16902987.
- ↑ "Lonsurf EPAR – Summary for the Public" (PDF). European Medicines Agency. April 2016.