Progestogen ester

A progestogen ester is an ester of a progestogen or progestin (a synthetic progestogen). The prototypical progestogen is progesterone, an endogenous sex hormone. Esterification is frequently employed to improve the pharmacokinetics of steroids, including oral bioavailability, lipophilicity, and half-life.[1] In addition, with intramuscular injection, steroid esters are often absorbed more slowly into the body, allowing for less frequent administration.[1] Many (though not all) steroid esters function as prodrugs. Esterification is particularly salient in the case of progesterone because progesterone itself shows very poor oral pharmacokinetics and is thus ineffective when taken orally.[2][3] Unmodified, it has a half-life of only 5 minutes, and is almost completely inactivated by the liver during first-pass metabolism.[3] Micronization, however, has allowed for progesterone to be effective orally, although oral micronized progesterone was not developed until recent years.[2]
Progestogen esters
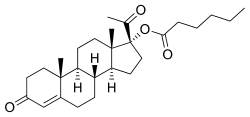
Estradiol was discovered in 1929,[4] and beginning in 1936, a variety of estradiol esters, such as estradiol benzoate and estradiol dipropionate, were introduced for clinical use.[4][5] Testosterone esters, such as testosterone propionate and testosterone phenylacetate, were also introduced around this time.[6] In contrast to estradiol and testosterone, progesterone proved more difficult to esterify.[7] In fact, esterification involves the replacement of a hydroxy group with an alkoxy group,[8] and unlike estradiol and testosterone, progesterone does not possess any hydroxy groups,[9] so it is actually not chemically possible to esterify progesterone itself. The first progestogen esters were not introduced until the mid-1950s,[4][7][10] and were esters of 17α-hydroxyprogesterone (which, unlike progesterone, has a hydroxy group available for esterification) rather than of progesterone; they included 17α-hydroxyprogesterone caproate (Delalutin, Proluton) and 17α-hydroxyprogesterone acetate (Prodrox).[2][10] The following quote of de Médicis Sajous et al. (1961) details the development of progestogen esters:[11]

Over a period of several years, many tens of thousands of dollars were invested by Upjohn in an effort to find an easily absorbed, orally active progesterone ester. The effort met with but limited success. One promising ester, [17α-hydroxyprogesterone acetate], marketed as Prodox, was found. It was more active by mouth than other progesterone preparations then on the market, but it was not so active orally as desired.To obtain a progestational drug with the wanted properties, it appeared necessary to alter the progesterone molecule itself. Beginning about 1957, Upjohn steroid chemists accordingly prepared a series of progesterones modified in the various ways that had been found to multiply the power of cortisone and hydrocortisone. One of the modifications — worked out by a team under Dr. John C. Babcock — was the attachment of a carbon atom and three hydrogen atoms — a methyl group — to carbon 6 in the first ring of the progesterone steroid nucleus. A similar modification had been the key step in creating Medrol, Upjohn's high-potency, antiinflammatory cortisone-type steroid. The new progestational agent was [6α-methyl-17α-hydroxyprogesterone acetate] or [medroxyprogesterone acetate], which Upjohn has trademarked Provera. It has proved to be the most potent progestational drug yet uncovered — hundreds of times more active orally than progesterone and, weight for weight, some fifty times more active by subcutaneous injection. Provera was placed on the market in 1959.
Medroxyprogesterone acetate (Provera) entered clinical use and became widely marketed, largely superseding the 17α-hydroxyprogesterone esters.[4] A variety of analogues of medroxyprogesterone acetate, such as chlormadinone acetate, cyproterone acetate, and megestrol acetate, were subsequently developed and introduced as well.[2][4][12] Progestogen esters of other groups of progestins have also been introduced, including the 19-norprogesterone derivatives gestonorone caproate, segesterone acetate (nestorone), nomegestrol acetate, and norgestomet (11β-methyl-17α-acetoxy-19-norprogesterone), and the 19-nortestosterone derivatives etynodiol diacetate, norethisterone acetate, norethisterone enanthate, and quingestanol acetate.
Although esters of steroidal androgens and estrogens are generally inactive themselves and act as prodrugs, not all progestogen esters do so. For instance, esters of 17α-hydroxyprogesterone derivatives, such as hydroxyprogesterone caproate and cyproterone acetate, are highly active themselves and are not prodrugs, forming little or none of their parent compounds (in the cases of the examples given, hydroxyprogesterone and cyproterone, respectively).[13][14] On the other hand, esters of 19-nortestosterone derivatives such as etynodiol diacetate, norethisterone acetate, norethisterone enanthate, and quingestanol acetate are all prodrugs.[15]
Progestogen ethers

Although it cannot be esterified, progesterone possesses a ketone group at the C3 position, and for this reason, it is possible to etherify it; that is, progesterone ethers are chemically possible. Quingestrone (Enol-Luteovis) is a progesterone ether (specifically, the 3-cyclopentyl ether of progesterone) that has been marketed in Italy as an oral contraceptive.[16][17] Quingestrone is a variant of progesterone with improved pharmacokinetics, including higher potency, oral activity, greater lipophilicity, and a longer half-life.[18][19][20][21][22] Two other progestogens, pentagestrone (never marketed) and pentagestrone acetate (Gestovis, Gestovister), are the 3-cyclopentyl enol ethers of 17α-hydroxyprogesterone and 17α-hydroxyprogesterone acetate, respectively.[3][16][3][23]
See also
References
- 1 2 Ian S. Fraser (1998). Estrogens and Progestogens in Clinical Practice. Churchill Livingstone. p. 13. ISBN 978-0-443-04706-0.
- 1 2 3 4 Roger Lobo; P.G. Crosignani; Rodolfo Paoletti (31 October 2002). Women’s Health and Menopause: New Strategies - Improved Quality of Life. Springer Science & Business Media. pp. 91–. ISBN 978-1-4020-7149-2.
- 1 2 3 4 Andrejus Korolkovas (16 August 1988). Essentials of Medicinal Chemistry. Wiley. p. 1021. ISBN 978-0-471-88356-2.
- 1 2 3 4 5 Enrique Ravina (11 January 2011). The Evolution of Drug Discovery: From Traditional Medicines to Modern Drugs. John Wiley & Sons. pp. 174–175, 194. ISBN 978-3-527-32669-3.
- ↑ Roche Review ... Hoffman-La Roche, and Roche-organon. 1944.
- ↑ Korenchevsky V, Dennison M, Eldridge M (1937). "The prolonged treatment of castrated and ovariectomized rats with testosterone propionate". Biochem. J. 31 (3): 475–85. doi:10.1042/bj0310475. PMC 1266958
 . PMID 16746360.
. PMID 16746360. - 1 2 Charles Eucharist de Medicis Sajous; Louis Theo de Me(dicis Sajous (1939). Analytic cyclopedia of practical medicine. Davis. p. 75.
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "esters".
- ↑ Nuclear Receptor Coregulators. Academic Press. 11 August 2004. pp. 69–. ISBN 978-0-08-052288-3.
- 1 2 Walter Sneader (23 June 2005). Drug Discovery: A History. John Wiley & Sons. pp. 204–. ISBN 978-0-471-89979-2.
- ↑ Leonard Engel (1961). Medicine Makers of Kalamazoo. McGraw-Hill. p. 125.
- ↑ Donna Shoupe (7 November 2007). The Handbook of Contraception: A Guide for Practical Management. Springer Science & Business Media. pp. 103–. ISBN 978-1-59745-150-5.
- ↑ Attardi BJ, Zeleznik A, Simhan H, Chiao JP, Mattison DR, Caritis SN (2007). "Comparison of progesterone and glucocorticoid receptor binding and stimulation of gene expression by progesterone, 17-alpha hydroxyprogesterone caproate, and related progestins". Am. J. Obstet. Gynecol. 197 (6): 599.e1–7. doi:10.1016/j.ajog.2007.05.024. PMC 2278032. PMID 18060946.
- ↑ Georg F. Weber (22 July 2015). Molecular Therapies of Cancer. Springer. pp. 316–. ISBN 978-3-319-13278-5.
- ↑ Stanley M. Roberts; Barry J. Price (1985). Medicinal chemistry: the role of organic chemistry in drug research. Academic Press. ISBN 978-0-12-589730-3.
- 1 2 J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 1058, 943. ISBN 978-1-4757-2085-3.
- ↑ International Planned Parenthood Federation. Medical Committee. Oral Advisory Group (1965). Handbook on oral contraception. Little, Brown. p. 18.
- ↑ Burton, Eunice R.; Wachtel, Erica G. (1967). "A CLINICAL TRIAL AND CYTOLOGICAL ASSESSMENT OF ENOL LUTEOVIS IN THE TREATMENT OF THREATENED AND RECURRENT ABORTION". BJOG: An International Journal of Obstetrics and Gynaecology. 74 (4): 533–536. doi:10.1111/j.1471-0528.1967.tb03986.x. ISSN 1470-0328.
- ↑ Charman, William N.; Porter, Christopher J.H. (1996). "Lipophilic prodrugs designed for intestinal lymphatic transport". Advanced Drug Delivery Reviews. 19 (2): 149–169. doi:10.1016/0169-409X(95)00105-G. ISSN 0169-409X.
- ↑ Joseph Bolivar De Lee (1965). The ... Year Book of Obstetrics and Gynecology. Year Book Publishers. p. 150.
- ↑ P. J. Bentley (1980). Endocrine Pharmacology: Physiological Basis and Therapeutic Applications. CUP Archive. pp. 274–. ISBN 978-0-521-22673-8.
- ↑ Current Medicine and Drugs. 1962.
Enol Luteovis (3 cyclo-pentyl enol ether of progesterone) is the only oral progestin producing pregnanediol as a metabolite. It is not very potent and probably carries very little risk of producing virilizing effects on a female foetus. Thus it is more closely related to progesterone than the other synthetic progestins.
- ↑ Camille Georges Wermuth (2 May 2011). The Practice of Medicinal Chemistry. Academic Press. pp. 731–. ISBN 978-0-08-056877-5.