Nucleic acid structure determination
Structure probing of nucleic acids is the process by which biochemical techniques are used to determine nucleic acid structure.[1] This analysis can be used to define the patterns which can infer the molecular structure, experimental analysis of molecular structure and function, and further understanding on development of smaller molecules for further biological research.[2] Structure probing analysis can be done through many different methods, which include chemical probing, hydroxyl radical probing, SHAPE, nucleotide analog interference mapping (NAIM), and in-line probing.
Physical methods
Nuclear magnetic resonance spectroscopy
Nucleic acid NMR is the use of NMR spectroscopy to obtain information about the structure and dynamics of nucleic acid molecules, such as DNA or RNA. As of 2003, nearly half of all known RNA structures had been determined by NMR spectroscopy.[3]
Nucleic acid NMR uses similar techniques as protein NMR, but has several differences. Nucleic acids have a smaller percentage of hydrogen atoms, which are the atoms usually observed in NMR, and because nucleic acid double helices are stiff and roughly linear, they do not fold back on themselves to give "long-range" correlations.[4] The types of NMR usually done with nucleic acids are 1H or proton NMR, 13C NMR, 15N NMR, and 31P NMR. Two-dimensional NMR methods are almost always used, such as correlation spectroscopy (COSY) and total coherence transfer spectroscopy (TOCSY) to detect through-bond nuclear couplings, and nuclear Overhauser effect spectroscopy (NOESY) to detect couplings between nuclei that are close to each other in space.[5]
Parameters taken from the spectrum, mainly NOESY cross-peaks and coupling constants, can be used to determine local structural features such as glycosidic bond angles, dihedral angles (using the Karplus equation), and sugar pucker conformations. For large-scale structure, these local parameters must be supplemented with other structural assumptions or models, because errors add up as the double helix is traversed, and unlike with proteins, the double helix does not have a compact interior and does not fold back upon itself. NMR is also useful for investigating nonstandard geometries such as bent helices, non-Watson–Crick basepairing, and coaxial stacking. It has been especially useful in probing the structure of natural RNA oligonucleotides, which tend to adopt complex conformations such as stem-loops and pseudoknots. NMR is also useful for probing the binding of nucleic acid molecules to other molecules, such as proteins or drugs, by seeing which resonances are shifted upon binding of the other molecule.[5]
RNA sequencing
While methods for each type of probing differ in steps, the probing of secondary structure involves certain steps in order to determine the structure. The first step involved is submitting the structural RNA to the probe of interest and incubating over a certain amount of time to allow the reaction to occur. The RNA is then transcribed using reverse transcriptase PCR, where this results in different lengths of bands due to the modification of the RNA at specific sites, which causes the reverse transcriptase to fall off.[6] These bands are then run on a gel with sequencing data determined from RNA sequencing.
Chemical methods
RNA chemical probing can involve many different chemicals which serve to modify specific bases at certain sites to show certain locations available for specific modification type.
Hydroxyl radical probing
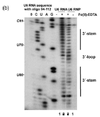
Probing with hydroxyl radicals involves an additional step, as hydroxyl radicals are short lived in solution they need to be generated. This can be done using H2O2, ascorbic acid, and Fe(II)-EDTA complex which is attached to the backbone through EDTA. The Fe(II) along with ascorbic acid generates hydroxyl radicals which then can react with the nucleic acid molecules.[7] Hydroxyl radical probing is often used in conjunction with chemical probing of nucleic acid molecules that are thought to associate with proteins, this is due to the modification done by hydroxyl radicals. Hydroxyl radicals attack the ribose/deoxyribose ring and this results in breaking of the phosphate backbone, which is independent of secondary structure, as all backbone is accessible, but is instead resultant of protein or tertiary structure protection.[7] Probing with hydroxyl radicals shows the protection of structured nucleic acids by the proteins thought to be associated or folding on itself, where cleavage again results in a band formed through gel electrophoresis (after RT-PCR in the case of RNA) that is shorter than the full nucleic acid depending upon where it is cleaved. In this case since the last nucleotide is not modified the band length is indicative of the base that was cleaved. When examining the gel produced by running the gels on a band the areas of various strength of protection where areas of stronger protection for hydroxyl radicals can be said to have tighter association with a protein, or if no protein associates with the nucleic acid it can be caused by the tertiary fold.[7][8]
DMS
Dimethyl sulfate, known as DMS, is a chemical that can be used to modify nucleic acids in order to determine secondary structure. DMS modifies certain bases through methylation. One set of methylation products that is used for RNA is the methylation of N1 adenosine and N3 of cytosine[9] which prevents the natural hydrogen bonds to form between bases modified by DMS. This enables modification sites to be detected by RT-PCR, as modified sites cannot be basepaired, which results in the reverse transcriptase to fall off and produce different band sizes. DMS may modify adenines and cytosines that are single stranded, base paired at the end of a helix, or in a base pair next to a GU pair.[10] Detection of these modifications is done by examination of a gel and looking at bands present. One thing of note is that since modification prevents the addition of nucleotides at the modification site each of the bands generated is shifted by one base down on the gel. Nucleotides may be protected from DMS modification by base pairing, tertiary contacts, or protein-RNA interactions. This can be used to begin to develop a model of secondary structure for the RNA molecule. Structure probing by DMS allows for detection of secondary structure changes due to binding of RNA molecules along with detecting changes in tertiary structure.[9]
DMS modification can also be used for DNA, for example in footprinting DNA-protein interactions.[11]
CMCT
1-cyclohexyl-(2-morpholinoethyl)carbodiimide metho-p-toluene sulfonate known as CMCT is another chemical that is used in structure probing. CMCT like DMS serves to modify the exposed bases of specific nucleotides, which are uridine, and to a smaller extent guanine.[12] CMCT reacts primarily with N3 of uridine and N1 of guanine modifying two sites responsible for hydrogen bonding on the bases.[9] Modification using CMCT is analogous in detection to DMS as modification prevents basepairing at specific nucleotides which are then detected using rt-PCR and running of the PCR products on an agarose gel as shown in Figure 1. Structure probing of RNA by CMCT indicates the presence of uridine and guanine in single stranded regions by accessibility to modification or the presence of uridine and guanine in double stranded regions by protection from CMCT, which is the absence of a band.[12] CMCT, like DMS can detect secondary and tertiary structure changes, but still has the same weaknesses as the method of modification is the same as DMS.
Kethoxal
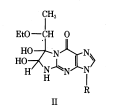
1,1-Dihydroxy-3-ethoxy-2-butanone, known as kethoxal, is used like DMS and CMCT, where treatment with kethoxal causes the modification of guanine, specifically altering the N1 and the exocyclic amino group simultaneously by covalent interaction.[14] The modification by kethoxal prevents single stranded guanine nucleotides to become modified, so that when reverse transcriptase reaches the modified guanine it falls off resulting in a band. After rt-PCR the products will be of different lengths where a band present indicates modification of a guanine base, and the absence of a band indicates that the base was not available for modification. Using this gel in combination with DMS and CMCT a model for structural RNAs can be formed through comparison between the gels which indicate protected and accessible positions in all the bases, which can be used to form a preliminary model.
SHAPE
Selective 2′-hydroxyl acylation analyzed by primer extension, or SHAPE, takes advantage of reagents that preferentially modify the backbone of RNA in structurally flexible regions.
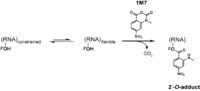
Reagents such as N-methylisotoic anhydride (NMIA) and 1-methyl-7-nitroisatoic anhydride (1M7) [15] react with the 2'-hydroxyl group to form adducts on the 2'-hydroxyl of the RNA backbone. Compared to the chemicals used in other RNA probing techniques, these reagents have the advantage of being largely unbiased to base identity, while remaining very sensitive to conformational dynamics. Nucleotides which are constrained (usually by base-pairing) show less adduct formation than nucleotides which are unpaired. Adduct formation is quantified for each nucleotide in a given RNA by extension of a complementary DNA primer with reverse transcriptase and comparison of the resulting fragments with those from an unmodified control.[16] SHAPE therefore reports on RNA structure at the individual nucleotide level. This data can be used as input to generate highly accurate secondary structure models.[17] SHAPE has been used to analyze diverse RNA structures, including that of an entire HIV-1 genome.[18] The best approach is to use a combination of chemical probing reagents and experimental data.[19] In SHAPE-Seq SHAPE is extended by bar-code based multiplexing combined with RNA-Seq and can be performed in a high-throughput fashion.[20]
In-line probing
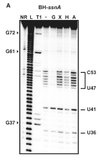
In-line probing does not involve treatment with any type of chemical or reagent to modify RNA structures. This type of probing assay uses the structure dependent cleavage of RNA; single stranded regions are more flexible and unstable and will degrade over time.[14] The process of in-line probing is often used to determine changes in structure due to ligand binding. Binding of a ligand can result in different cleavage patterns. The process of in-line probing involves incubation of structural or functional RNAs over a long period of time. This period can be several days, but varies in each experiment. The incubated products are then run on a gel to visualize the bands. This experiment is often done using two different conditions: 1) with ligand and 2) in the absence of ligand.[21] Cleavage results in shorter band lengths and is indicative of areas that are not basepaired, as basepaired regions tend to be less sensitive to spontaneous cleavage.[14] In-line probing is a functional assay that can be used to determine structural changes in RNA in response to ligand binding. It can directly show the change in flexibility and binding of regions of RNA in response to a ligand, as well as compare that response to analogous ligands. This assay is commonly used in dynamic studies, specifically when examining riboswitches[14]
Nucleotide analog interference mapping
Nucleotide analog interference mapping (NAIM) is the process of using nucleotide analogs, molecules that are similar in some ways to nucleotides but lack function, to determine the importance of a functional group at each location of an RNA molecule.[14] The process of NAIM is to insert a single nucleotide analog into a unique site. This can be done by transcribing a short RNA using T7 RNA polymerase, then synthesizing a short oligonucleotide containing the analog in a specific position, then ligating them together on the DNA template using a ligase.[22] The nucleotide analogs are tagged with a phosphorothioate, the active members of the RNA population are then distinguished from the inactive members, the inactive members then have the phosphorothioate tag removed and the analog sites are identified using gel electrophoresis and autoradiography.[22] This indicates a functionally important nucleotide, as cleavage of the phosphorothioate by iodine results in an RNA that is cleaved at the site of the nucleotide analog insert. By running these truncated RNA molecules on a gel, the nucleotide of interest can be identified against a sequencing experiment[23] Site directed incorporation results indicate positions of importance where when running on a gel, functional RNAs that have the analog incorporated at that position will have a band present, but if the analog results in non-functionality, when the functional RNA molecules are run on a gel there will be no band corresponding to that position on the gel.[24] This process can be used to evaluate an entire area, where analogs are placed in site specific locations, differing by a single nucleotide, then when functional RNAs are isolated and run on a gel, all areas where bands are produced indicate non-essential nucleotides, but areas where bands are absent from the functional RNA indicate that inserting a nucleotide analog in that position caused the RNA molecule to become non-functional[22]
References
- ↑ Teunissen AWM (1979). RNA Structure Probing: Biochemical structure analysis of autoimmune-related RNA molecules. pp. 1–27. ISBN 90-901323-4-1.
- ↑ Pace NR, Thomas BC, Woese CR (1999). Probing RNA Structure, Function, and History by Comparative Analysis. Cold Spring Harbor Laboratory Press. pp. 113–117. ISBN 0-87969-589-7.
- ↑ Fürtig B, Richter C, Wöhnert J, Schwalbe H (October 2003). "NMR spectroscopy of RNA". Chembiochem. 4 (10): 936–62. doi:10.1002/cbic.200300700. PMID 14523911.
- ↑ Addess, Kenneth J.; Feigon, Juli (1996). "Introduction to 1H NMR Spectroscopy of DNA". In Hecht, Sidney M. Bioorganic Chemistry: Nucleic Acids. New York: Oxford University Press. ISBN 0-19-508467-5.
- 1 2 Wemmer, David (2000). "Chapter 5: Structure and Dynamics by NMR". In Bloomfield, Victor A.; Crothers, Donald M.; Tinoco, Ignacio. Nucleic acids: Structures, Properties, and Functions. Sausalito, California: University Science Books. ISBN 0-935702-49-0.
- ↑ Yu E, Fabris D (2003). "Direct probing of RNA structures and RNA-Protein interactions in the HIV-1 packaging signal by chemical modification and electrospray ionization fourier transform mass spectrometry". J. Mol. Biol. 330 (2): 211–223. doi:10.1016/S0022-2836(03)00589-8. PMID 12823962.
- 1 2 3 4 Karaduman R, Fabrizio P, Hartmuth K, Urlaub H, Luhrmann R (2006). "RNA structure and RNA-protein interactions in purified yeast U6 snRNPs". J. Mol. Biol. 356 (5): 1248–1262. doi:10.1016/j.jmb.2005.12.013. PMID 16410014.
- ↑ Tullius, T. D.; Dombroski, B. A. (1986). "Hydroxyl radical "footprinting": high-resolution information about DNA-protein contacts and application to lambda repressor and Cro protein". Proceedings of the National Academy of Sciences. 83 (15): 5469–5473. Bibcode:1986PNAS...83.5469T. doi:10.1073/pnas.83.15.5469. PMC 386308
 . PMID 3090544.
. PMID 3090544. - 1 2 3 Tijerina P, Mohr S, Russell R (2007). "DMS footprinting of structured RNAs and RNA-protein complexes". Nat Protoc. 2 (10): 2608–23. doi:10.1038/nprot.2007.380. PMC 2701642. PMID 17948004.
- ↑ Mathews, DH; Disney, MD; Childs, JL; Schroeder, SJ; Zuker, M; Turner DH (2004). "Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure". Proceedings of the National Academy of Sciences. 101: 7287–7292. Bibcode:2004PNAS..101.7287M. doi:10.1073/pnas.0401799101. PMC 409911. PMID 15123812.
- ↑ Albert S. Baldwin Jr.; Marjorie Oettinger & Kevin Struhl (1996). "Unit 12.3: Methylation and Uracil Interference Assays for Analysis of Protein-DNA Interactions". Current Protocols in Molecular Biology. Wiley. doi:10.1002/0471142727.mb1203s36.
- 1 2 Fritz JJ, Lewin A, Hauswirth W, Agarwal A, Grant M, Shaw L (2002). "Development of hammerhead ribozymes to modulate endogenous gene expression for functional studies". Methods. 28 (2): 276–285. doi:10.1016/S1046-2023(02)00233-5. PMID 12413427.
- ↑ Noller HF, Chaires JB (1972). "Functional modification of 16S ribosomal RNA by kethoxal". Proc. Natl. Acad. Sci. USA. 69 (11): 3115–3118. Bibcode:1972PNAS...69.3115N. doi:10.1073/pnas.69.11.3115. PMC 389716. PMID 4564202.
- 1 2 3 4 5 Gopinath SCB (2009). "Mapping of RNA-protein interactions". Analytica Chimica Acta. 636 (2): 117–128. doi:10.1016/j.aca.2009.01.052. PMID 19264161.
- ↑ Mortimer SA, Weeks KM (2007). "A Fast-Acting Reagent for Accurate Analysis of RNA Secondary and Tertiary Structure by SHAPE Chemistry". J Am Chem Soc. 129 (14): 4144–45. doi:10.1021/ja0704028. PMID 17367143.
- ↑ Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM (2005). "RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE)". J Am Chem Soc. 127 (12): 4223–31. doi:10.1021/ja043822v. PMID 15783204.
- ↑ Deigan KE, Li TW, Mathews DH, Weeks KM (2009). "Accurate SHAPE-directed RNA structure determination". Proc Natl Acad Sci USA. 106 (1): 97–102. Bibcode:2009PNAS..106...97D. doi:10.1073/pnas.0806929106. PMC 2629221. PMID 19109441.
- ↑ Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, Burch CL, Weeks KM (2009). "Architecture and secondary structure of an entire HIV-1 RNA genome". Nature. 460 (7256): 711–6. Bibcode:2009Natur.460..711W. doi:10.1038/nature08237. PMC 2724670. PMID 19661910.
- ↑ Wipapat Kladwang; Christopher C. VanLang; Pablo Cordero; Rhiju Das (7 Sep 2011). "Understanding the errors of SHAPE-directed RNA structure modeling". arXiv:1103.5458.
- ↑ Lucks JB, Mortimer SA, Trapnell C, Luo S, Aviran S, Schroth GP, Pachter L, Doudna JA, Arkin AP (2011). "Multiplexed RNA structure characterization with selective 2'-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq).". Proc Natl Acad Sci USA. 108 (27): 11063–8. doi:10.1073/pnas.1106501108. PMC 3131332. PMID 21642531.
- 1 2 Muhlbacher J, Lafontaine DA (2007). "Ligand recognition determinants of guanine riboswitches". Nucleic Acids Research. 35 (16): 5568–5580. doi:10.1093/nar/gkm572. PMC 2018637. PMID 17704135.
- 1 2 3 Ryder SP, Strobel SA (1999). "Nucleotide Analog Interference Mapping". Methods: A Comparison to Methods in Enzymology. 18: 38–50. doi:10.1006/meth.1999.0755.
- ↑ Waldsich C (2008). "Dissecting RNA folding by nucleotide analog interference mapping (NAIM)". Nature Protocols. 3 (5): 811–823. doi:10.1038/nprot.2008.45. PMC 2873565. PMID 18451789.
- ↑ Strobel SA, Shetty K (1997). "Defining the chemical groups essential for Tetrahymena group I intron function by nucleotide analog interference mapping". Proc. Natl. Acad. Sci. USA. 94 (7): 2903–2908. Bibcode:1997PNAS...94.2903S. doi:10.1073/pnas.94.7.2903. PMC 20295. PMID 9096319.