Reductions with diimide
Reductions with diimide are a chemical reactions that convert unsaturated organic compounds to reduced alkane products. In the process, diimide (N
2H
2) is oxidized to dinitrogen.[1]
Introduction
In 1929, the conversion of oleic acid to stearic acid in the presence of hydrazine was observed.[2] The short-lived intermediate diimide was not implicated in this reductive process until the 1960s. Since that time, several methods of generating transient amounts of diimide have been developed.[3][4] In the presence of unpolarized alkenes, alkynes or allenes, diimide is converted into dinitrogen with reduction (net addition of dihydrogen) of the unsaturated functionality. Diimide formation is the rate-limiting step of the process, and a concerted mechanism involving cis-diimide has been proposed.[5] This reduction represents a metal-free alternative to catalytic hydrogenation reductions, and does not lead to the cleavage of sensitive O–O and N–O bonds.
(1)
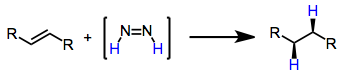
Mechanism and Stereochemistry
Prevailing Mechanism
Diimide reductions result in the syn addition of dihydrogen to alkenes and alkynes. This observation has led to the proposal that the mechanism involves concerted hydrogen transfer from cis-diimide to the substrate. The cis isomer is the less stable of the two; however, acid catalysis may speed up equilibration of the trans and cis isomers.[5]
(2)
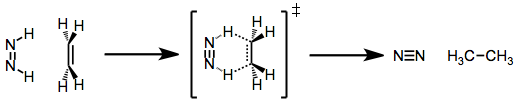
Diimide is typically generated either through the oxidation of hydrazine or the decarboxylation of potassium azodicarboxylate. Kinetic experiments suggest that regardless of its method of generation, the formation of diimide is rate-limiting. The transition state of the hydrogen transfer step is likely early; however, high stereoselectivity has been obtained in many reductions of chiral alkenes.[6]
(3)
The order of reactivity of unsaturated substrates is: alkynes, allenes > terminal or strained alkenes > substituted alkenes. Trans alkenes react more rapidly than cis alkenes in general. The reactivity difference between alkynes and alkenes is usually not great enough to isolate intermediate alkenes; however, alkenes can be isolated from allene reductions. Diimide reduces symmetrical double bonds i.e.,C=C. N=N, O=O etc. unsymmetrical double bonds can not be reduced
Scope and Limitations
Diimide is most effective at reducing unpolarized carbon-carbon double or triple bonds. In reactions with other unsaturated systems, disproportionation of diimide to nitrogen gas and hydrazine is a competing process that significantly degrades the reducing agent. Many groups that are ordinarily sensitive to reductive conditions, including peroxides, are not affected by the conditions of diimide reductions.[7]
(4)
Diimide will selectively reduce less substituted double bonds under some conditions. Discrimination between terminal and disubstituted double bonds is often low, however.
(5)
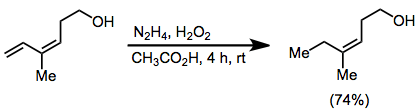
Allenes are reduced to the more highly substituted alkene in the presence of diimide, although yields are low.[8]
(6)
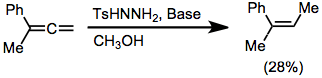
Iodoalkynes represent an exception to the rule that alkenes cannot be obtained from alkynes. After diimide reduction of iodoalkynes, cis-iodoalkenes may be isolated in good yield.[9]
(7)
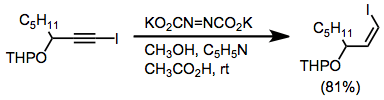
Recently, diimide has been generated catalytically through the oxidation of hydrazine by a flavin-based organocatalyst. This system selectively reduces terminal double bonds.[10]
(8)
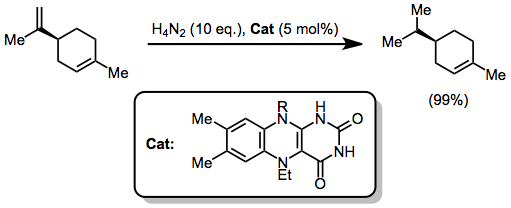
In general, diimide does not efficiently reduce polarized double bonds; however, a limited number of examples do exist in the literature. Aromatic aldehydes are reduced by diimide generated through the decarboxylation of potassium azodicarboxylate.[11]
Comparison with Other Methods
Reductions of carbon-carbon double and triple bonds are most commonly accomplished through catalytic hydrogenation:[12](9)
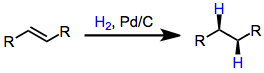
However, diimide reduction offers the advantages that the handling of gaseous hydrogen is unnecessary and removal of catalysts and byproducts (one of which is gaseous dinitrogen) is straightforward. Hydrogenolysis side reactions do not occur during diimide reductions, and N–O and O–O bonds are not affected by the reaction conditions. On the other hand, diimide reductions often require long reaction times, and reductions of highly substituted or polarized double bonds are sluggish.
In addition, an excess of the reagent used to generate diimide (e.g. dipotassium azodicarboxylate) is required for hydrogenation because of the two competing processes of disproportionation (to N
2H
4 and N
2) and decomposition (to N
2 and H
2) that the liberated diimide can also undergo.[13][14] Unfortunately, this means that in the case of alkyne reduction, over-reduction to the alkane can occur resulting in diminished yields where the cis alkene is the desired product.[14]
Experimental Conditions and Procedure
Typical Conditions
A variety of methods for the generation of diimide exist. The most synthetically useful methods are:
- Oxidation of hydrazine with oxygen, in the presence of a copper(II) catalyst and/or a carboxylic acid
- Decarboxylation of dipotassium azodicarboxylate in the presence of an acid
- Thermal decomposition of sulfonylhydrazides
Procedures (particularly those employing air as an oxidant) are typically straightforward and do not require special handling techniques.
Example Procedure[15]
(10)
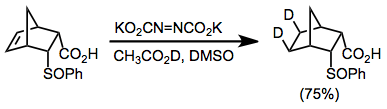
Acetic acid-O-d (1.2 g) was slowly added dropwise into a solution of the carboxylic acid starting material (200 mg, 1 mmol) and dipotassium azodicarboxylate (400 mg, 2.5 mmol) in DMSO (7 mL). After stirring 4 hours at room temperature the solution was diluted with brine and extracted with pentane. The pentane layer was dried and evaporated to afford 160 mg (75%) of 5-exo-6-exo-dideuterio-3-endo-phenylsulfinylbicyclo[2.2.1]heptane-2-endo-carboxylic acid: mp 183–184°. 1H NMR (DMSO-d6): δ 7.32 (s, 5H) 4.37 (m, 1H), 4.17 (m, 1H), 3.68 (m, 1H), 3.48 (m, 1H), 2.60 (m, 2H), 1.49 (br s, 2H).
References
- ↑ Pasto, D. J.; Taylor, R. T. Org. React. 1991, 40, 91. doi:10.1002/0471264180.or040.02
- ↑ Hanuš, J.; Voríšek, J. Collect. Czech. Chem. Commun. 1929, 1, 223.
- ↑ Aylward, F.; Sawistowska, M. Chem. Ind. (London), 1962, 484.
- ↑ Cusack, J.; Reese, B.; Risius, C.; Roozpeikar, B. Tetrahedron 1976, 32, 2157.
- 1 2 Spears, Jr., L. G.; Hutchinson, J. S. J. Chem. Phys. 1988, 88, 240.
- ↑ Corey, J.; Pasto, J.; Mock, L. J. Am. Chem. Soc. 1961, 83, 2957.
- ↑ Adam, W.; Eggelte, J. J. Org. Chem. 1977, 42, 3987.
- ↑ Mori, K.; Ohki, M.; Sato, A.; Matsui, M. Tetrahedron 1972, 28, 3739.
- ↑ Luethy, C.; Konstantin, P.; Untch, J. G. J. Am. Chem. Soc. 1978, 100, 6211.
- ↑ Smit, C.; Fraaije, M.; Minnaard, A. J. Org. Chem. 2008, 73, 9482.
- ↑ Curry, C.; Uff, C.; Ward, D. J. Chem. Soc. C, 1967, 1120.
- ↑ Birch, A.; Williamson, D. Org. React. 1976, 24, 1.
- ↑ Hünig, S.; Müller, H.,R.; Thier, W. Angew. Chem. Int. Ed. 1965, 4, 271-382.
- 1 2 Miller, C. E. J. Chem. Ed. 1965, 42, 254-259.
- ↑ Annunziata, R.; Fornasier, R.; Montanari, F. J. Org. Chem. 1974, 39, 3195.
16.A. Gangadhar, T. Chandrasekhara Rao, R. Subbarao, G. Lakshminarayana, Journal of the American Oil Chemists Society October 1989, Volume 66, Issue 10, pp 1507-1508 17. A. Gangadhar, R. Subbarao, G. Lakshminarayana, Journal of the American Oil Chemists Society July 1984, Volume 61, Issue 7, pp 1239-1241