Seaborgium
| General properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name, symbol | seaborgium, Sg | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pronunciation |
see-BOR-gee-əm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Seaborgium in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 106 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group, block | group 6, d-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Element category | transition metal | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight (Ar) | [269] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Rn] 5f14 6d4 7s2 [1] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
per shell | 2, 8, 18, 32, 32, 12, 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase | solid (predicted)[2] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density near r.t. | 35.0 g/cm3 (predicted)[1][3] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | 6, (5), (4), (3), 0[1][3] (parenthesized oxidation states are predictions) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
1st: 752.6 kJ/mol 2nd: 1732.9 kJ/mol 3rd: 2483.5 kJ/mol (more) (all but first estimated)[1] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic radius | empirical: 132 pm (predicted)[1] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | 143 pm (estimated)[4] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Miscellanea | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure |
body-centered cubic (bcc) 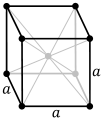
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 54038-81-2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Naming | after Glenn T. Seaborg | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery | Lawrence Berkeley National Laboratory (1974) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Most stable isotopes of seaborgium | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Seaborgium is a chemical element with symbol Sg and atomic number 106. It is named after the American nuclear chemist Glenn T. Seaborg. It is one of only two elements named after a living person at the time of naming, the other being oganesson. It is a synthetic element (an element that can be created in a laboratory but is not found in nature) and radioactive; the most stable known isotope, 269Sg, has a half-life of approximately 3.1 minutes.
In the periodic table of the elements, it is a d-block transactinide element. It is a member of the 7th period and belongs to the group 6 elements as the fourth member of the 6d series of transition metals. Chemistry experiments have confirmed that seaborgium behaves as the heavier homologue to tungsten in group 6. The chemical properties of seaborgium are characterized only partly, but they compare well with the chemistry of the other group 6 elements.
In 1974, a few atoms of seaborgium were produced in laboratories in the former Soviet Union and in the United States. The priority of the discovery and therefore the naming of the element was disputed between Soviet and American scientists, and it was not until 1997 that International Union of Pure and Applied Chemistry (IUPAC) established seaborgium as the official name for the element.
History

Two groups claimed discovery of the element. Evidence of element 106 was first reported in 1974 by a Russian research team in Dubna led by Yuri Oganessian, in which targets of lead-208 and lead-207 were bombarded with accelerated ions of chromium-54. In total, fifty-one spontaneous fission events were observed with a half-life between four and ten milliseconds. After having ruled out nucleon transfer reactions as a cause for these activities, the team concluded that the most likely cause of the activities was the spontaneous fission of isotopes of element 106. The isotope in question was first suggested to be seaborgium-259, but was later corrected to seaborgium-260.[5]
A few months later in 1974, researchers at the University of California, Berkeley, including Glenn T. Seaborg and Albert Ghiorso, also synthesised the element by bombarding a californium-249 target with oxygen-18 ions, using equipment similar to that which had been used for the synthesis of element 104 five years earlier, observing at least seventy alpha decays, seemingly from the isotope seaborgium-263m with a half-life of 0.9±0.2 seconds. The alpha daughter rutherfordium-259 and granddaughter nobelium-255 had previously been synthesised and the properties observed here matched with those previously known, as did the intensity of their production. The cross-section of the reaction observed, 0.3 nanobarns, also agreed well with theoretical predictions. These bolstered the assignment of the alpha decay events to seaborgium-263m.[5]
A dispute thus arose from the initial competing claims of discovery, though unlike the case of the synthetic elements up to element 105, neither team of discoverers chose to announce proposed names for the new elements, thus averting an element naming controversy temporarily. The dispute on discovery, however, dragged on until 1992, when the IUPAC/IUPAP Transfermium Working Group (TWG), formed to put an end to the controversy by making conclusions regarding discovery claims for elements 101 to 112, concluded that the Soviet synthesis of seaborgium-260 was not convincing enough, "lacking as it is in yield curves and angular selection results", whereas the American synthesis of seaborgium-263 was convincing due to its being firmly anchored to known daughter nuclei. As such, the TWG recognised the Berkeley team as official discoverers in their 1993 report.[5]
Seaborg had previously suggested to the TWG that if Berkeley was recognised as the official discoverer of elements 104 and 105, they might propose the name kurchatovium (symbol Kt) for element 106 to honour the Dubna team, which had proposed this name for element 104 after Igor Kurchatov, the former head of the Soviet nuclear research programme. However, due to the worsening relations between the competing teams after the publication of the TWG report (because the Berkeley team vehemently disagreed with the TWG's conclusions, especially regarding element 104), this proposal was dropped from consideration by the Berkeley team.[6] After being recognised as official discoverers, the Berkeley team started deciding on a name in earnest:[7]
...we were given credit for the discovery and the accompanying right to name the new element. The eight members of the Ghiorso group suggested a wide range of names honoring Isaac Newton, Thomas Edison, Leonardo da Vinci, Ferdinand Magellan, the mythical Ulysses, George Washington, and Finland, the native land of a member of the team. There was no focus and no front-runner for a long period.
Then one day Al [Ghiorso] walked into my office and asked what I thought of naming element 106 "seaborgium." I was floored.[7]— Glenn T. Seaborg
The name seaborgium and symbol Sg were announced at the 207th national meeting of the American Chemical Society in March 1994 by Kenneth Hulet, who was on the Berkeley team that discovered the element.[7] However, IUPAC resolved in August 1994 that an element could not be named after a living person, and Seaborg was still alive at the time. Thus, in September 1994, IUPAC recommended a set of names in which the names proposed by the three laboratories (the third being the GSI Helmholtz Centre for Heavy Ion Research in Darmstadt, Germany) with competing claims to the discovery for elements 104 to 109 were shifted to various other elements, in which rutherfordium (Rf), the Berkeley proposal for element 104, was shifted to element 106, with seaborgium being dropped entirely as a name.[6]
| Atomic number | Systematic | American | Russian | German | Compromise 92 | IUPAC 94 | ACS 94 | IUPAC 95 | IUPAC 97 | Present |
|---|---|---|---|---|---|---|---|---|---|---|
| 101 | unnilunium | mendelevium | — | — | mendelevium | mendelevium | mendelevium | mendelevium | mendelevium | mendelevium |
| 102 | unnilbium | nobelium | joliotium | — | joliotium | nobelium | nobelium | flerovium | nobelium | nobelium |
| 103 | unniltrium | lawrencium | rutherfordium | — | lawrencium | lawrencium | lawrencium | lawrencium | lawrencium | lawrencium |
| 104 | unnilquadium | rutherfordium | kurchatovium | — | meitnerium | dubnium | rutherfordium | dubnium | rutherfordium | rutherfordium |
| 105 | unnilpentium | hahnium | nielsbohrium | — | kurchatovium | joliotium | hahnium | joliotium | dubnium | dubnium |
| 106 | unnilhexium | seaborgium | — | — | rutherfordium | rutherfordium | seaborgium | seaborgium | seaborgium | seaborgium |
| 107 | unnilseptium | — | — | nielsbohrium | nielsbohrium | bohrium | nielsbohrium | nielsbohrium | bohrium | bohrium |
| 108 | unniloctium | — | — | hassium | hassium | hahnium | hassium | hahnium | hassium | hassium |
| 109 | unnilennium | — | — | meitnerium | hahnium | meitnerium | meitnerium | meitnerium | meitnerium | meitnerium |
| 110 | ununnilium | hahnium | becquerelium | darmstadtium | — | — | — | — | — | darmstadtium |
| 111 | unununium | — | — | roentgenium | — | — | — | — | — | roentgenium |
| 112 | ununbium | — | — | copernicium | — | — | — | — | — | copernicium |
This decision ignited a firestorm of worldwide protest for disregarding the historic discoverer's right to name new elements, and against the new retroactive rule against naming elements after living persons; the American Chemical Society stood firmly behind the name seaborgium for element 106, together with all the other American and German naming proposals for elements 104 to 109, approving these names for its journals in defiance of IUPAC.[6] At first, IUPAC defended itself, with an American member of its committee writing: "Discoverers don't have a right to name an element. They have a right to suggest a name. And, of course, we didn't infringe on that at all." However, Seaborg responded:[7]
This would be the first time in history that the acknowledged and uncontested discoverers of an element are denied the privilege of naming it.[7]— Glenn T. Seaborg
Bowing to public pressure, IUPAC proposed a different compromise in August 1995, in which the name seaborgium was reinstated for element 106 in exchange for the removal of all the other American proposals, which met an even worse response. Finally, IUPAC rescinded these previous compromises and made a final, new recommendation in August 1997, in which the American and German proposals for elements 104 to 109 were all adopted, including seaborgium for element 106, with the single exception of element 105, named dubnium to recognise the contributions of the Dubna team to the experimental procedures of transactinide synthesis. This list was finally accepted by the American Chemical Society, which wrote:[6]
In the interest of international harmony, the Committee reluctantly accepted the name 'dubnium' for element 105 in place of 'hahnium' [the American proposal], which has had long-standing use in literature. We are pleased to note that 'seaborgium' is now the internationally approved name for element 106.[6]— American Chemical Society
Seaborg commented regarding the naming:[7]
I am, needless to say, proud that U.S. chemists recommended that element 106, which is placed under tungsten (74), be called 'seaborgium.' I was looking forward to the day when chemical investigators will refer to such compounds as seaborgous chloride, seaborgic nitrate, and perhaps, sodium seaborgate.
This is the greatest honor ever bestowed upon me—even better, I think, than winning the Nobel Prize.[lower-alpha 1] Future students of chemistry, in learning about the periodic table, may have reason to ask why the element was named for me, and thereby learn more about my work.[7]— Glenn T. Seaborg
Seaborg died two years later, on 25 February 1999, at the age of 86.[7]
Isotopes
| Isotope | Half-life [9][10] | Decay mode[9][10] | Discovery year | Reaction |
|---|---|---|---|---|
| 258Sg | 3 ms | SF | 1994 | 209Bi(51V,2n) |
| 259Sg | 600 ms | α | 1985 | 207Pb(54Cr,2n) |
| 260Sg | 4 ms | SF, α | 1985 | 208Pb(54Cr,2n) |
| 261Sg | 200 ms | α, EC, SF | 1985 | 208Pb(54Cr,n) |
| 261mSg | 92 μs | IT | 2009 | 208Pb(54Cr,n) |
| 262Sg | 7 ms | SF, α | 2001 | 270Ds(—,2α) |
| 263Sg | 1 s | α | 1994 | 271Ds(—,2α) |
| 263mSg | 120 ms | α, SF | 1974 | 249Cf(18O,4n) |
| 264Sg | 37 ms | SF | 2006 | 238U(34Si,4n) |
| 265Sg | 8 s | α | 1993 | 248Cm(22Ne,5n) |
| 265mSg | 16.2 s | α | 1993 | 248Cm(22Ne,5n) |
| 266Sg | 360 ms | SF | 2004 | 270Hs(—,2α) |
| 267Sg | 1.4 min | SF, α | 2004 | 271Hs(—,2α) |
| 268Sg | 30? s | ? | unknown | — |
| 269Sg | 3.1 min | α | 2010 | 285Fl(—,4α) |
| 270Sg | 10? min | ? | unknown | — |
| 271Sg | 2.4 min | α | 2003 | 287Fl(—,4α) |
| 272Sg | 1? h | ? | unknown | — |
| 273Sg | 1? min | ? | unknown | — |
Super-heavy elements such as seaborgium are produced by bombarding lighter elements in particle accelerators that induces fusion reactions. Whereas most of the isotopes of seaborgium can be synthesized directly this way, some heavier ones have only been observed as decay products of elements with higher atomic numbers.[11]
Depending on the energies involved, the former are separated into "hot" and "cold". In hot fusion reactions, very light, high-energy projectiles are accelerated toward very heavy targets (actinides), giving rise to compound nuclei at high excitation energy (~40–50 MeV) that may either fission or evaporate several (3 to 5) neutrons.[11] In cold fusion reactions, the produced fused nuclei have a relatively low excitation energy (~10–20 MeV), which decreases the probability that these products will undergo fission reactions. As the fused nuclei cool to the ground state, they require emission of only one or two neutrons, and thus, allows for the generation of more neutron-rich products.[12] The latter is a distinct concept from that of where nuclear fusion claimed to be achieved at room temperature conditions (see cold fusion).[13]
Seaborgium has no stable or naturally-occurring isotopes. Several radioactive isotopes have been synthesized in the laboratory, either by fusing two atoms or by observing the decay of heavier elements. Twelve different isotopes of seaborgium have been reported with atomic masses 258–267, 269, and 271, three of which, seaborgium-261, 263, and 265, have known metastable states. All of these decay only through alpha decay and spontaneous fission, with the single exception of seaborgium-261 that can also undergo electron capture to dubnium-261.[9]
There is a trend toward increasing half-lives for the heavier isotopes; thus the heaviest three known isotopes, 267Sg, 269Sg, and 271Sg, are also the longest-lived, having half-lives in minutes. Some other isotopes in this region are predicted to have comparable or even longer half-lives, with the longest-lived predicted isotope being 272Sg which is expected to have a half-life of about an hour. Additionally, 263Sg, 265Sg, 265mSg, as well as the predicted 268Sg have or should have half-lives measured in seconds. All the remaining isotopes have half-lives measured in milliseconds, with the exception of the shortest-lived isotope, 261mSg, with a half-life of only 92 microseconds.[9]
The proton-rich isotopes from 258Sg to 261Sg were directly produced by cold fusion; all heavier isotopes were produced from the repeated alpha decay of the heavier elements hassium, darmstadtium, and flerovium, with the exceptions of the isotopes 263mSg, 264Sg, 265Sg, and 265mSg, which were directly produced by hot fusion through irradiation of actinide targets. The twelve isotopes of seaborgium have half-lives ranging from 92 microseconds for 261Sg to 3.1 minutes for 269Sg.[9]
Properties
Physical
Seaborgium is expected to be a solid under normal conditions and assume a body-centered cubic crystal structure, similar to its lighter congener tungsten.[2] It should be a very heavy metal with a density of around 35.0 g/cm3, which would be the fourth-highest of any of the 118 known elements, lower than only bohrium (37.1 g/cm3), meitnerium (37.4 g/cm3) and hassium (41 g/cm3), the three following elements in the periodic table.[1] In comparison, the densest known element that has had its density measured, osmium, has a density of only 22.61 g/cm3. This results from seaborgium's high atomic weight, the lanthanide and actinide contractions, and relativistic effects, although production of enough seaborgium to measure this quantity would be impractical, and the sample would quickly decay.[1]
Chemical
Seaborgium is the fourth member of the 6d series of transition metals and the heaviest member of group 6 in the periodic table, below chromium, molybdenum, and tungsten. All the members of the group form a diversity of oxoanions. They readily portray their group oxidation state of +6, although this is highly oxidising in the case of chromium, and this state becomes more and more stable to reduction as the group is descended: indeed, tungsten is the last of the 5d transition metals where all four 5d electrons participate in metallic bonding.[14] As such, seaborgium should have +6 as its most stable oxidation state, both in the gas phase and in aqueous solution, and this is the only oxidation state that is experimentally known for it; the +5 and +4 states should be less stable and the +3 state, the most common for chromium, would be the least stable for seaborgium.[1] Experimental chemical investigation has been hampered due to the need to produce seaborgium one atom at a time, its short half-life, and the resulting necessary harshness of the experimental conditions.[15]
This stabilisation of the highest oxidation state occurs in the early 6d elements because of the similarity between the energies of the 6d and 7s orbitals, since the 7s orbitals are relativistically stabilised and the 6d orbitals are relativistically destabilised. This effect is so large in the seventh period that seaborgium is expected to lose 6d electrons before 7s electrons (Sg, [Rn]5f146d47s2; Sg+, [Rn]5f146d37s2; Sg2+, [Rn]5f146d37s1; Sg4+, [Rn]5f146d2; Sg6+, [Rn]5f14). Because of the great destabilisation of the 7s orbital, Sg4+ should be even more unstable than W4+ and should be very readily oxidised to Sg6+. The predicted ionic radius of the hexacoordinate Sg6+ ion is 65 pm, while the predicted atomic radius of seaborgium is 128 pm. Nevertheless, the stability of the highest oxidation state is still expected to decrease as Lr3+ > Rf4+ > Db5+ > Sg6+. Some predicted standard reduction potentials for seaborgium ions in aqueous acidic solution are as follows:[1]
| 2 SgO3 + 2 H+ + 2 e− | ⇌ Sg2O5 + H2O | E0 = −0.046 V |
| Sg2O5 + 2 H+ + 2 e− | ⇌ 2 SgO2 + H2O | E0 = +0.11 V |
| SgO2 + 4 H+ + e− | ⇌ Sg3+ + 2 H2O | E0 = −1.34 V |
| Sg3+ + e− | ⇌ Sg2+ | E0 = −0.11 V |
| Sg3+ + 3 e− | ⇌ Sg | E0 = +0.27 V |
Seaborgium should form a very volatile hexafluoride (SgF6) as well as a moderately volatile hexachloride (SgCl6), pentachloride (SgCl5), and oxychlorides SgO2Cl2 and SgOCl4.[3] SgO2Cl2 is expected to be the most stable of the seaborgium oxychlorides and to be the least volatile of the group 6 oxychlorides, with the sequence MoO2Cl2 > WO2Cl2 > SgO2Cl2.[1]
The volatile seaborgium(VI) compounds SgCl6 and SgOCl4 are expected to be unstable to decomposition to seaborgium(V) compounds at high temperatures, analogous to MoCl6 and MoOCl4; this should not happen for SgO2Cl2 due to the much higher energy gap between the highest occupied and lowest unoccupied molecular orbitals, despite the similar Sg–Cl bond strengths (similarly to molybdenum and tungsten).[16] Thus, in the first experimental chemical studies of seaborgium in 1995 and 1996, seaborgium atoms were produced in the reaction 248Cm(22Ne,4n)266Sg, thermalised, and reacted with an O2/HCl mixture. The adsorption properties of the resulting oxychloride were measured and compared with those of molybdenum and tungsten compounds. The results indicated that seaborgium formed a volatile oxychloride akin to those of the other group 6 elements, and confirmed the decreasing trend of oxychloride volatility down group 6:
- Sg + O
2 + 2 HCl → SgO
2Cl
2 + H
2
In 2001, a team continued the study of the gas phase chemistry of seaborgium by reacting the element with O2 in a H2O environment. In a manner similar to the formation of the oxychloride, the results of the experiment indicated the formation of seaborgium oxide hydroxide, a reaction well known among the lighter group 6 homologues as well as the pseudohomologue uranium.[17]
- 2 Sg + 3 O
2 → 2 SgO
3 - SgO
3 + H
2O → SgO
2(OH)
2
Molybdenum and tungsten are very similar to each other and show important differences to the smaller chromium, and seaborgium is expected to follow the chemistry of tungsten and molybdenum quite closely, forming an even greater variety of oxoanions, the simplest among them being seaborgate, SgO2−
4, which would form from the rapid hydrolysis of Sg(H
2O)6+
6, although this would take place less readily than with molybdenum and tungsten as expected from seaborgium's greater size. Seaborgium should hydrolyse less readily than tungsten in hydrofluoric acid at low concentrations, but more readily at high concentrations, also forming complexes such as SgO3F− and SgOF−
5: complex formation competes with hydrolysis in hydrofluoric acid.[1] These predictions have largely been confirmed. In experiments conducted in 1997 and 1998, seaborgium was eluted from cation-exchange resin using a HNO3/HF solution, most likely as neutral SgO2F2 or the anionic complex ion [SgO2F3]− rather than SgO2−
4. In contrast, in 0.1 M nitric acid, seaborgium does not elute, unlike molybdenum and tungsten, indicating that the hydrolysis of [Sg(H2O)6]6+ only proceeds as far as the cationic complex [Sg(OH)4(H2O)]2+ or [Sg(OH)3(H2O)2]+, while that of molybdenum and tungsten proceeds to neutral [MO2(OH)2)].[1]
The only other oxidation state known for seaborgium other than the group oxidation state of +6 is the zero oxidation state. Similarly to its three lighter congeners, forming chromium hexacarbonyl, molybdenum hexacarbonyl, and tungsten hexacarbonyl, seaborgium has been shown in 2014 to also form seaborgium hexacarbonyl, Sg(CO)6. Like its molybdenum and tungsten homologues, seaborgium hexacarbonyl is a volatile compound that reacts readily with silicon dioxide.[15]
Notes
- ↑ Seaborg had in fact previously won the 1951 Nobel Prize in Chemistry together with Edwin McMillan for "their discoveries in the chemistry of the first transuranium elements".[8]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 Hoffman, Darleane C.; Lee, Diana M.; Pershina, Valeria (2006). "Transactinides and the future elements". In Morss; Edelstein, Norman M.; Fuger, Jean. The Chemistry of the Actinide and Transactinide Elements (3rd ed.). Dordrecht, The Netherlands: Springer Science+Business Media. ISBN 1-4020-3555-1.
- 1 2 3 Östlin, A.; Vitos, L. (2011). "First-principles calculation of the structural stability of 6d transition metals". Physical Review B. 84 (11). Bibcode:2011PhRvB..84k3104O. doi:10.1103/PhysRevB.84.113104.
- 1 2 3 Fricke, Burkhard (1975). "Superheavy elements: a prediction of their chemical and physical properties". Recent Impact of Physics on Inorganic Chemistry. 21: 89–144. doi:10.1007/BFb0116498. Retrieved 4 October 2013.
- ↑ Chemical Data. Seaborgium – Sg, Royal Chemical Society
- 1 2 3 Barber, R. C.; Greenwood, N.N.; Hrynkiewicz, A.Z.; Jeannin, Y.P.; Lefort, M.; Sakai, M.; Ulehla, I.; Wapstra, A.P.; Wilkinson, D.H. (1993). "Discovery of the transfermium elements. Part II: Introduction to discovery profiles. Part III: Discovery profiles of the transfermium elements". Pure and Applied Chemistry. 65 (8): 1757. doi:10.1351/pac199365081757.
- 1 2 3 4 5 6 Hoffman, D.C., Ghiorso, A., Seaborg, G. T. The Transuranium People: The Inside Story, (2000), 369–399
- 1 2 3 4 5 6 7 8 "106 Seaborgium". Elements.vanderkrogt.net. Retrieved 2008-09-12.
- ↑ "The Nobel Prize in Chemistry 1951". Nobel Foundation. Retrieved August 26, 2012.
- 1 2 3 4 5 Sonzogni, Alejandro. "Interactive Chart of Nuclides". National Nuclear Data Center: Brookhaven National Laboratory. Retrieved 2008-06-06.
- 1 2 Gray, Theodore (2002–2010). "The Photographic Periodic Table of the Elements". periodictable.com. Retrieved 16 November 2012.
- 1 2 Barber, Robert C.; Gäggeler, Heinz W.; Karol, Paul J.; Nakahara, Hiromichi; Vardaci, Emanuele; Vogt, Erich (2009). "Discovery of the element with atomic number 112 (IUPAC Technical Report)". Pure and Applied Chemistry. 81 (7): 1331. doi:10.1351/PAC-REP-08-03-05.
- ↑ Armbruster, Peter & Munzenberg, Gottfried (1989). "Creating superheavy elements". Scientific American. 34: 36–42.
- ↑ Fleischmann, Martin; Pons, Stanley (1989). "Electrochemically induced nuclear fusion of deuterium". Journal of Electroanalytical Chemistry and Interfacial Electrochemistry. 261 (2): 301–308. doi:10.1016/0022-0728(89)80006-3.
- ↑ Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. pp. 1002–39. ISBN 0-08-037941-9.
- 1 2 Even, J.; Yakushev, A.; Dullmann, C. E.; Haba, H.; Asai, M.; Sato, T. K.; Brand, H.; Di Nitto, A.; Eichler, R.; Fan, F. L.; Hartmann, W.; Huang, M.; Jager, E.; Kaji, D.; Kanaya, J.; Kaneya, Y.; Khuyagbaatar, J.; Kindler, B.; Kratz, J. V.; Krier, J.; Kudou, Y.; Kurz, N.; Lommel, B.; Miyashita, S.; Morimoto, K.; Morita, K.; Murakami, M.; Nagame, Y.; Nitsche, H.; et al. (2014). "Synthesis and detection of a seaborgium carbonyl complex". Science. 345 (6203): 1491. doi:10.1126/science.1255720. PMID 25237098.
- ↑ Kratz, J. V. (2003). "Critical evaluation of the chemical properties of the transactinide elements (IUPAC Technical Report)" (PDF). Pure and Applied Chemistry. 75 (1): 103. doi:10.1351/pac200375010103.
- ↑ Huebener, S.; Taut, S.; Vahle, A.; Dressler, R.; Eichler, B.; Gäggeler, H. W.; Jost, D.T.; Piguet, D.; et al. (2001). "Physico-chemical characterization of seaborgium as oxide hydroxide" (PDF). Radiochim. Acta. 89 (11–12_2001): 737–741. doi:10.1524/ract.2001.89.11-12.737.
External links
| Wikimedia Commons has media related to Seaborgium. |
- Chemistry in its element podcast (MP3) from the Royal Society of Chemistry's Chemistry World: Seaborgium
- Seaborgium at The Periodic Table of Videos (University of Nottingham)
- WebElements.com – Seaborgium
| Periodic table (Large cells) | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||||||||||||||||
| 1 | H | He | |||||||||||||||||||||||||||||||
| 2 | Li | Be | B | C | N | O | F | Ne | |||||||||||||||||||||||||
| 3 | Na | Mg | Al | Si | P | S | Cl | Ar | |||||||||||||||||||||||||
| 4 | K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr | |||||||||||||||
| 5 | Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe | |||||||||||||||
| 6 | Cs | Ba | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn | |
| 7 | Fr | Ra | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og | |
|
| |||||||||||||||||||||||||||||||||