Selenoxide elimination
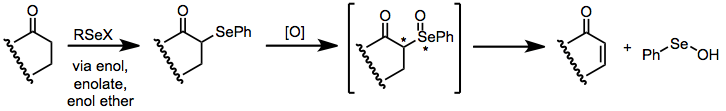
Selenoxide elimination (also called α-selenation)[1] is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues.[2]
Mechanism and stereochemistry
After the development of sulfoxide elimination as an effective method for generating carbon–carbon double bonds,[3] it was discovered that selenoxides undergo a similar process, albeit much more rapidly. Most selenoxides decompose to the corresponding alkenes at temperatures between −50 and 40 °C. Evidence suggests that the elimination is syn; however, epimerization at both carbon and selenium (both of which are stereogenic) may occur during the reaction. As selenoxides can be readily prepared from nucleophilic carbonyl derivatives (enols and enolates),[4] selenoxide elimination has grown into a general method for the preparation of α,β-unsaturated carbonyl compounds.
(1)
Mechanism
Elimination of selenoxides takes place through an intramolecular syn elimination pathway. The carbon–hydrogen and carbon–selenium bonds are co-planar in the transition state.[5]
(2)
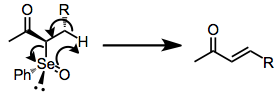
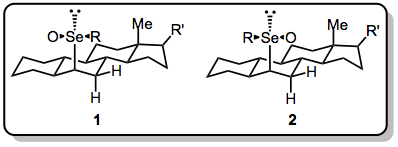
The reaction is highly trans-selective when acyclic α-phenylseleno carbonyl compounds are employed. Formation of conjugated double bonds is favored. Endocyclic double bonds tend to predominate over exocyclic ones, unless no syn hydrogen is available in the ring. Selenium in these reactions is almost always stereogenic, and the effect of epimerization at selenium (which is acid-catalyzed and occurs readily) on the elimination reaction is nearly unknown. In one example, separation and warming of selenoxides 1 and 2 revealed that 2 decomposes at 0 °C, while 1, which presumably has more difficulty accessing the necessary syn conformation for elimination, is stable to 5 °C.[6]
(3)
Scope and limitations
Selenylating and oxidizing reagents
α-Selenylation of carbonyl compounds can be accomplished with electrophilic or nucleophilic selenylating reagents. Usually, simple phenylseleno compounds are used in elimination reactions; although 2-nitrophenylselenides react more quickly, they are more expensive to prepare, and phenylselenides typically react in minutes. Electrophilic selenylating reagents can be used in conjunction with enols, enolates, or enol ethers. Phenylselenating reagents include:
- Diphenyl diselenide
- Benzeneselenyl chloride
- Benzeneselenyl bromide
- Benzeneselinyl chloride
- Sodium benzeneselenolate
- Trimethylsilyl phenyl selenide
The most common oxidizing agent employed is hydrogen peroxide (H2O2).[7] It is sometimes used in excess, to overcome catalytic decomposition of H2O2 by selenium; however, undesired oxidation of starting material has been observed under these conditions. Oxidation of products (via the Baeyer-Villiger reaction, for instance) has also been observed.[8]
(4)
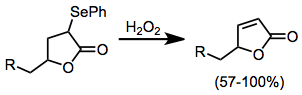
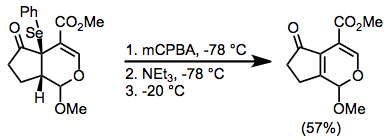
For substrates whose product olefins are sensitive to oxidation, meta-Chloroperoxybenzoic acid (mCPBA) can be employed as an oxidant. It oxidizes selenides below the temperature at which they decompose to alkenes; thus, all oxidant is consumed before elimination begins. Buffering with an amine base is necessary before warming to avoid acid-mediated side reactions.[9]
(5)
Ozone, which gives only dioxygen as a byproduct after oxidation, is used to oxidize selenides when special conditions are required for thermolysis or extreme care is necessary during workup. Quinones can be synthesized from the corresponding cyclic unsaturated carbonyl compounds using this method.[10]
(6)
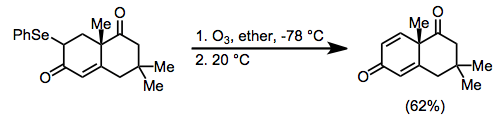
Substrates
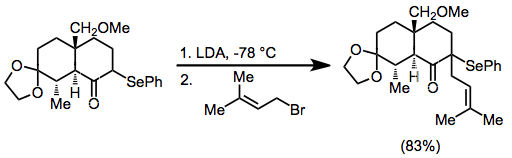
α-Phenylseleno aldehydes, which are usually prepared from the corresponding enol ethers, are usually oxidized with mCPBA or ozone, as hydrogen peroxide causes over-oxidation. α-Phenylseleno ketones can be prepared by kinetically controlled enolate formation and trapping with an electrophilic selenylating reagent such as benzeneselenyl chloride. A second deprotonation, forming a selenium-substituted enolate, allows alkylation or hydroxyalkylation of these substrates.[11]
(7)
Base-sensitive substrates may be selenylated under acid-catalyzed conditions (as enols) using benzeneselenyl chloride. Hydrochloric acid generated during the selenylation of transient enol catalyzes tautomerization.[12]
(8)
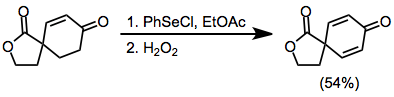
The seleno-Pummerer reaction is a significant side reaction that may occur under conditions when acid is present.[13] Protonation of the selenoxide intermediate, followed by elimination of hydroxide and hydrolysis, leads to α-dicarbonyl compounds. The reaction is not a problem for more electron-rich carbonyls—generally, fewer side reactions are observed in eliminations of esters and amides.[13]
(9)
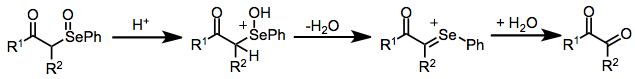
A second significant side reaction in reactions of ketones and aldehydes is selenylation of the intermediate selenoxide. This process leads to elimination products retaining a carbon-selenium bond,[14] and is more difficult to prevent than the seleno-Pummerer reaction. Tertiary selenoxides, which are unable to undergo enolization, do not react further with selenium electrophiles.
(10)
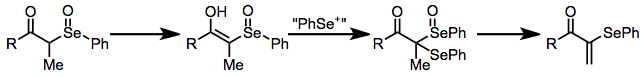
Comparison with other methods
Analogous sulfoxide eliminations are generally harder to implement than selenoxide eliminations. Formation of the carbon–sulfur bond is usually accomplished with highly reactive sulfenyl chlorides, which must be prepared for immediate use. However, sulfoxides are more stable than the corresponding selenoxides, and elimination is usually carried out as a distinct operation. This allows thermolysis conditions to be optimized (although the high temperatures required may cause other thermal processes). In addition, sulfoxides may be carried through multiple synthetic steps before elimination is carried out.[15]
(11)

The combination of silyl enol ethers with palladium(II) acetate (Pd(OAc)2), the Saegusa oxidation, gives enones. However, the reaction requires stoichiometric amounts of Pd(OAc)2 and thus is not amenable to large-scale synthesis.[16] Catalytic variants have been developed.[17]
(12)
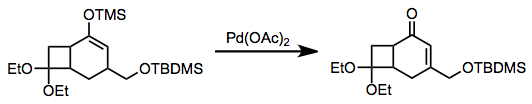
For β-dicarbonyl compounds, DDQ can be used as an oxidizing agent in the synthesis of enediones. Additionally, some specialized systems give better yields upon DDQ oxidation.[18]
(13)
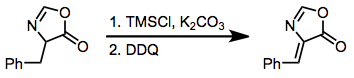
See also
References
- ↑ T. W. Graham Solomons; Craig B. Fryhle (2008). "Chapter 17 Aldehydes and Ketones-Part II: Enols and Enolate Ions". Organic Chemistry, 9th Edition. Wiley. p. 759. ISBN 978-0-470-16982-7.
- ↑ Reich, H. J.; Wollowitz, S. Org. React. 1993, 44, 1. doi:10.1002/0471264180.or044.01
- ↑ Emersohjgjvbn, David W.; Craig, Arthur P.; Potts, Irvin W. (January 1967). "Pyrolysis of unsymmetrical dialkyl sulfoxides. Rates of alkene formation and composition of the gaseous products". The Journal of Organic Chemistry. 32 (1): 102–105. doi:10.1021/jo01277a026.
- ↑ Sharpless, K. B.; Lauer, R. F.; Teranishi, A. Y. (September 1973). "Electrophilic and nucleophilic organoselenium reagents. New routes to .alpha.,.beta.-unsaturated carbonyl compounds". Journal of the American Chemical Society. 95 (18): 6137–6139. doi:10.1021/ja00799a062.
- ↑ Sharpless, K.B.; Young, M.W.; Lauer, R.F. (January 1973). "Reactions of selenoxides: Thermal -elimination and H218O exchange.". Tetrahedron Letters. 14 (22): 1979–1982. doi:10.1016/S0040-4039(01)96098-8.
- ↑ Jones, D. Neville; Mundy, D.; Whitehouse, R. D. (1970). "Steroidal selenoxides diastereoisomeric at selenium; syn-elimination, absolute configuration, and optical rotatory dispersion characteristics". Journal of the Chemical Society D: Chemical Communications (2): 86. doi:10.1039/C29700000086.
- ↑ Figueredo, Marta; Font, Josep; Virgili, Albert (January 1987). "Studies on structurally simple α,β-butenolides". Tetrahedron. 43 (8): 1881–1886. doi:10.1016/S0040-4020(01)81500-3.
- ↑ Vargas, David; Fronczek, Frank R.; Fischer, Nikolaus H.; Hostettmann, Kurt (January 1986). "The Chemistry of Confertiflorin and the Molecular Structure of Confertiflorin and Allodesacetylconfertiflorin, Two Molluscicidal Sesquiterpene Lactones". Journal of Natural Products. 49 (1): 133–138. doi:10.1021/np50043a017.
- ↑ Callant, Paul; Ongena, Raymond; Vandewalle, Maurits (January 1981). "Iridoids : Novel total synthesis of (±)- isoiridomyrmecin and of (±)-verbenalol". Tetrahedron. 37 (11): 2085–2089. doi:10.1016/S0040-4020(01)97962-1.
- ↑ Waring, Anthony John; Zaidi, Javid Hussain (1985). "Synthesis of a 4-acylcyclohexa-2,5-dienone: 3,4-dihydro-3,3,8a-trimethyl-naphthalene-1,6(2H,8aH)-dione". Journal of the Chemical Society, Perkin Transactions 1: 631. doi:10.1039/P19850000631.
- ↑ Solomon, Mark; Hoekstra, William; Zima, George; Liotta, Dennis (October 1988). "The case favoring direct C-alkylation of heteroatom-substituted enolates". The Journal of Organic Chemistry. 53 (21): 5058–5062. doi:10.1021/jo00256a029.
- ↑ Plieninger, Hans; Gramlich, Walter (May 1978). "Darstellung von Cyclohexadienonen, III: Synthese und Eigenschaften von (Phenylseleno)cyclohexenonen und deren Überführung in Cyclohexadienone". Chemische Berichte (in German). 111 (5): 1944–1957. doi:10.1002/cber.19781110528.
- 1 2 Reich, Hans J.; Renga, James M.; Reich, Ieva L. (July 1974). "Organoselenium chemistry. Conversion of cyclic ketones and .beta.-dicarbonyl compounds to enones". The Journal of Organic Chemistry. 39 (14): 2133–2135. doi:10.1021/jo00928a042.
- ↑ Reich, Hans J.; Renga, James M.; Reich, Ieva L. (September 1975). "Organoselenium chemistry. Conversion of ketones to enones by selenoxide syn elimination". Journal of the American Chemical Society. 97 (19): 5434–5447. doi:10.1021/ja00852a019.
- ↑ Wakamatsu, Takeshi; Yamada, Satoshi; Ban, Yoshio (1986). "Synthesis of dl-Vermiculine via Control of Olefin Formation". HETEROCYCLES. 24 (2): 309. doi:10.3987/R-1986-02-0309.
- ↑ Ito, Yoshihiko; Hirao, Toshikazu; Saegusa, Takeo (March 1978). "Synthesis of .alpha.,.beta.-unsaturated carbonyl compounds by palladium(II)-catalyzed dehydrosilylation of silyl enol ethers". The Journal of Organic Chemistry. 43 (5): 1011–1013. doi:10.1021/jo00399a052.
- ↑ Tsuji, Jro; Takahashi, Kazuhiko; Minami, Ichiro; Shimizu, Isao (January 1984). "Palladium-catalyzed preparation of α-allyl esters and α,β-unsaturated esters from saturated esters via their silyl acetals". Tetrahedron Letters. 25 (42): 4783–4786. doi:10.1016/S0040-4039(01)81518-5.
- ↑ Lott, Richard S.; Breitholle, Edward G.; Stammer, Charles H. (March 1980). "Azlactone oxidation". The Journal of Organic Chemistry. 45 (6): 1151–1153. doi:10.1021/jo01294a046.