Type II topoisomerase

Type II topoisomerases cut both strands of the DNA helix simultaneously in order to manage DNA tangles and supercoils. They use the hydrolysis of ATP, unlike Type I topoisomerase. In this process, these enzymes change the linking number of circular DNA by ±2.
Function
Once cut, the ends of the DNA are separated, and a second DNA duplex is passed through the break. Following passage, the cut DNA is religated. This reaction allows type II topoisomerases to increase or decrease the linking number of a DNA loop by 2 units, and it promotes chromosome disentanglement. Reactions involving the increase in supercoiling require two molecules of ATP. For example, DNA gyrase, a type II topoisomerase observed in E. coli and most other prokaryotes, introduces negative supercoils and decreases the linking number by 2. Gyrase is also able to remove knots from the bacterial chromosome. Along with gyrase, most prokaryotes also contain a second type IIA topoisomerase, termed topoisomerase IV. Gyrase and topoisomerase IV differ by their C-terminal domains, which is believed to dictate substrate specificity and functionality for these two enzymes. Footprinting indicates that gyrase, which forms a 140-base-pair footprint and wraps DNA, introduces negative supercoils, while topoisomerase IV, which forms a 28-base-pair footprint, does not wrap DNA.
Eukaryotic type II topoisomerase cannot introduce supercoils; it can only relax them.
The roles of type IIB topoisomerases are less understood. Unlike type IIA topoisomerases, type IIB topoisomerases cannot simplify DNA topology (see below), but they share several structural features with type IIA topoisomerases.
Topology simplification
Type IIA topoisomerases are essential in the separation of entangled daughter strands during replication. This function is believed to be performed by topoisomerase II in eukaryotes and by topoisomerase IV in prokaryotes. Failure to separate these strands leads to cell death. Type IIA topoisomerases have the special ability to relax DNA to a state below that of thermodynamic equilibrium, a feature unlike type IA, IB, and IIB topoisomerases. This ability, known as topology simplification, was first identified by Rybenkov et al. (Science 1997). The hydrolysis of ATP drives this simplification, but a clear molecular mechanism for this simplification is still lacking. Several models to explain this phenomenon have been proposed, including two models that rely on the ability of type IIA topoisomerases to recognize bent DNA duplexes (Vologodskiy, Proceedings of the National Academy of Sciences 1999). Biochemistry, electron microscopy, and recent structures of topoisomerase II bound to DNA reveal that type IIA topoisomerases bind at the apices of DNA, supporting this model.
Classification
There are two subclasses of type II topoisomerases, type IIA and IIB.
- Type IIA topoisomerases include the enzymes DNA gyrase, eukaryotic topoisomerase II (topo II), and bacterial topoisomerase IV (topo IV). These enzymes span all domains of life and are essential for function.
- Type IIB topoisomerases are structurally and biochemically distinct, and comprise a single family member, topoisomerase VI (topo VI). Type IIB topoisomerases are found in archaea and some higher plants.
Some organisms have two isoforms of topoisomerase II: alpha and beta. In cancers, the topoisomerase II-alpha is highly expressed in highly proliferating cells. In certain cancers, such as peripheral nerve sheath tumors, high expression of its encoded protein is also associated to poor patient survival.
The two classes of topoisomerases possess a similar strand passage mechanism and domain structure (see below), however they also have several important differences. Type IIA topoisomerases form double-stranded breaks with four-base pair overhangs, while type IIB topoisomerases form double-stranded breaks with two base overhangs (Buhler, Lebbink, Bocs, Ladenstein, and Forterre, Journal of Biological Chemistry 2001). In addition, type IIA topoisomerases are able to simplify DNA topology (Rybenkov Science 1997), while type IIB topoisomerases do not (Corbett Journal of Molecular Biology, 2006).
Structure of type IIA topoisomerases
| DNA Topoisomerase IIA | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
DNA topoisomerase IIA dimer, Human | |||||||||
| Identifiers | |||||||||
| EC number | 5.99.1.3 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
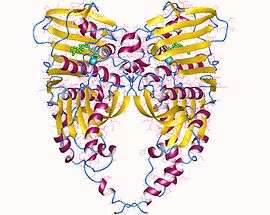
Type IIA topoisomerases consist of several key motifs: an N-terminal GHKL ATPase domain (for gyrase, Hsp, kinase and MutL), a Toprim domain (sometimes called a Rossmann fold), which exists in both type II topoisomerases, type IA topoisomerases, and bacterial primase (DnaG), a central DNA-binding core (which structurally forms a heart-shaped structure), and a variable C-terminal domain.
Eukaryotic type II topoisomerases are homodimers (A2), while prokaryotic type II topoisomerases are heterodimers (A2B2). Prokaryotes have the ATPase domain and the Toprim fold on one polypeptide, while the DNA cleavage core and the CTD lies on a second polypeptide. For gyrase, the first polypeptide is called GyrB and the second polypeptide is called GyrA. For topo IV, the first polypeptide is called ParE and the second polypeptide is called ParC.
The structures of the N-terminal ATPase domain of gyrase (Wigley, Davies, Dodson, Maxwell, and Dodson, Nature 1991) and yeast topoisomerase II (Classen and Berger, Proceedings of the National Academy of Sciences, 2003, PDB ID=1PVG) have been solved in complex with AMPPNP (an ATP analogue), showing that two ATPase domains dimerize to form a closed conformation. For gyrase, the structure has a substantial hole in the middle, which is presumed to accommodate the T-segment.
Linking the ATPase domain to the Toprim fold is a helical element known as the transducer domain. This domain is thought to communicate the nucleotide state of the ATPase domain to the rest of the protein. Modifications to this domain affect topoisomerase activity, and structural work done by the Verdine group shows that the ATP state affects the orientation of the transducer domain (Journal of Biological Chemistry, 2006).
The central core of the protein contains a Toprim fold and a DNA-binding core that contains a winged helix domain (WHD), often referred to as a CAP domain, since it was first identified to resemble the WHD of catabolite activator protein. The catalytic tyrosine lies on this WHD. The Toprim fold is a Rossmann fold that contains three invariant acidic residues that coordinate magnesium ions involved in DNA cleavage and DNA religation (Avarind, Leipe, Konin, Nucleic Acids Research 1998). The structure of the Toprim fold and DNA-binding core of yeast topoisomerase II was first solved by Berger and Wang (Nature 1996, PDB ID = 1BGW), and the first gyrase DNA-binding core was solved by Morais Cabral et al. (Nature 1997, PDB ID = 1AB4). The structure solved by Berger revealed important insights into the function of the enzyme. The DNA-binding core consists of the WHD, which leads to a tower domain. A coiled-coil region leads to a C-terminal domain that forms the main dimer interface for this crystal state (often termed the C-gate). While the original topoisomerase II structure shows a situation where the WHDs are separated by a large distance, the structure of gyrase shows a closed conformation, where the WHD close.

The topoisomerase II core was later solved in new conformations, including one by Fass et al. (Nature Structure Biology 1999, PDB ID = 1BJT) and one by Dong et al. (Nature 2007, PDB ID = 2RGR). The Fass structure shows that the Toprim domain is flexible and that this flexibility can allow the Toprim domain to coordinate with the WHD to form a competent cleavage complex. This was eventually substantiated by the Dong et al. structure that was solved in the presence of DNA. This last structure showed that the Toprim domain and the WHD formed a cleavage complex very similar to that of the type IA topoisomerases and indicated how DNA-binding and cleavage could be uncoupled, and the structure showed that DNA was bent by ~150 degrees through an invariant isoleucine (in topoisomerase II it is I833 and in gyrase it is I172). This mechanism of bending resembles closely that of integration host factor (IHF) and HU, two architectural proteins in bacteria. In addition, while the previous structures of the DNA-binding core had the C-gate closed, this structure captured the gate open, a key step in the two-gate mechanism (see below).
More recently, several structures of the DNA-bound structure have been solved in an attempt to understand both the chemical mechanism for DNA cleavage and the structural basis for inhibition of topoisomerase by antibacterial poisons.
The C-terminal region of the prokaryotic topoisomerases has been solved for multiple species. The first structure of a C-terminal domain of gyrase was solved by Corbett et al. (Proceedings of the National Academy of Sciences, 2004, PDB ID = 1SUU), and the C-terminal domain of topoisomerase IV was solved by Corbett et al. (Journal of Molecular Biology, 2006, PDB ID = 1zvt and 1zvu). The structures formed a novel beta barrel, which bends DNA by wrapping the nucleic acid around itself. The bending of DNA by gyrase has been proposed as a key mechanism in the ability of gyrase to introduce negative supercoils into the DNA. This is consistent with footprinting data that shows that gyrase has a 140-base-pair footprint. Both gyrase and topoisomerase IV CTDs bend DNA, but only gyrase introduces negative supercoils.
Unlike the function of the C-terminal domain of prokaryotic topoisomerases, the function of the C-terminal region of eukaryotic topoisomerase II is still not clear. Studies have suggested that this region is regulated by phosphorylation and this modulates topoisomerase activity, however more research needs to be done to investigate this.
Structures of type IIB topoisomerases
The organization of type IIB topoisomerases are similar to that of type IIAs, except that all type IIBs have two genes and form heterodimers. One gene, termed topo VI-B (since it resembles gyrB), contains the ATPase domain, a H2TH domain, and the transducer domain. The second gene, termed topo VI-A, contains the WHD and the Toprim domain.
The ATPase domain of topo VI B was solved in multiple nucleotide states (Corbett and Berger, EMBO J 2003). It closely resembles that of the GHKL domain of topo II and MutL and shows that the nucleotide state (ADP versus ATP) effects the orientation of the transducer domain (pdb ID= 1MU5 and 1MX0).
The structure of topo VI-A was solved by Bergerat et al. (Nature 1997), showing that the HTH and Toprim fold had a novel conformation compared with that of topo IIA.
A recent structure of the topo VI A/B complex was solved, showing an open and closed conformation, two states that are predicted in the two-gate mechanism (see below). These structures, of which one is an X-ray crystal structure and the other is a Small-Angle X-ray Scattering (SAXS) reconstruction, show that the ATPase domain can be either open or closed (Corbett, Benedetti, Berger Nature Structure Molecular Biology, 2007, PDB ID = 2Q2E).

Strand passage mechanism of type II topoisomerases
Type IIA topoisomerase operates through a "two-gate" mechanism (though this is a historical notation), a mechanism supported by biochemistry (Roca and Wang) as well as by structural work (Berger and Wang) (see above).
A strand of DNA, called the gate, or G-segment, is bound by a central DNA-binding gate (DNA-gate). A second strand of DNA, called the transport, or T-segment, is captured by the dimerization of the N-terminal ATPase domain (the ATPase-gate) when two molecules of ATP bind. Hydrolysis of ATP and release of an inorganic phosphate leads to the cleavage of the G-segment, as the catalytic tyrosines form a covalent phosphotyrosine bond with the 5' end of the DNA. This creates a four-base overhang and a double-stranded break in the G-segment. As the DNA-binding gate separates, the T-segment is transferred through the G-segment. The G-segment is sealed, leading to the C-terminal gate (or C-gate) to open, allowing for the release of the T-segment. Release of product ADP leads to a reset of the system, and allows a second T-segment to be captured.
Type IIB topoisomerases operate through a similar fashion, except that the protein forms a two-base overhang in the G-segment and the C-terminal gate is completely missing.
DNA cleavage mechanism of type IIA topoisomerases
In the strand passage mechanism, the cleavage of DNA is key to allow the T-segment to transfer through the G-segment. The mechanism of DNA cleavage by type IIA topoisomerases has recently been the focus of many biochemical and structural biology studies.
Catenation
Catenation is the process by which two circular DNA strands are linked together like chain links. This occurs after DNA replication, where two single strands are catenated and can still replicate but cannot separate into the two daughter cells. As type II topoisomerses break a double strand, they can fix this state (type I topoisomerases could do this only if there were already a single-strand nick), and the correct chromosome number can remain in daughter cells. Linear DNA in eukaryotes is so long they can be thought of as being without ends; type II topoisomerases are needed for the same reason.
Inhibition
Small molecules that target type II topoisomerase are divided into two classes: inhibitors and poisons.
- Inhibitors of type II topoisomerase include HU-331, ICRF-187, ICRF-193, and mitindomide. These molecules work by inhibiting the ATPase activity by acting as noncompetitive inhibitors of ATP. This has been shown through structural studies (Classen et al. Proceedings of the National Academy of Sciences, 2005) and biochemical studies performed by the Lindsley group.
- Poisons of type II topoisomerases include etoposide, novobiocin, quinolones (including ciprofloxacin), and teniposide. These small molecules target the DNA-protein complex. Some of these molecules lead to increased cleavage, whereas others, such as etoposide, inhibit religation.
The experimental antitumor drug m-AMSA (4'-(9'-acridinylamino)methanesulfon-m-anisidide) also inhibits type 2 topoisomerase.[1]
Topoisomerase poisons have been extensively used as both anticancer and antibacterial therapies. While antibacterial compounds such as ciprofloxacin target bacterial gyrase, they fail to inhibit eukaryotic type IIA topoisomerases. In addition, drug-resistant bacteria often have a point mutation in gyrase (Serine79Alanine in E. coli) that renders quinolones ineffective. Recent structural studies have led to the discovery of a compound that no longer relies on this residue and, therefore, has efficacy against drug-resistant bacteria.
References
- ↑ Willmore E, de Caux S, Sunter NJ, et al. (2004). "A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia". Blood. 103 (12): 4659–65. doi:10.1182/blood-2003-07-2527. PMID 15010369.
- Wang, J.C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002 Jun;3(6):430-40.
External links
| Wikimedia Commons has media related to Type II DNA topoisomerase. |
- DNA Topoisomerases, Type II at the US National Library of Medicine Medical Subject Headings (MeSH)