Astroblastoma
| Astroblastoma | |
|---|---|
|
Micrograph of an astroblastoma showing the characteristic nuclear pervivascular pseudorosette. H&E stain. | |
| Classification and external resources | |
| Specialty | oncology |
| ICD-10 | C71.9 |
| ICD-9-CM | 191.9 |
| ICD-O | 9430/3 |
| DiseasesDB | None |
| MeSH | D018302 |
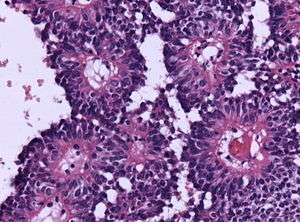
Astroblastoma is a rare glial tumor derived from the astroblast, a type of cell that closely resembles spongioblastoma and astrocytes.[1] Astroblastoma cells are most likely found in the supratentorial region of the brain that houses the cerebrum, an area responsible for all voluntary movements in the body.[2] It also occurs significantly in the frontal lobe, parietal lobe, and temporal lobe, areas where movement, language creation, memory perception, and environmental surroundings are expressed. These tumors can be present in major brain areas not associated with the main cerebral hemispheres, including the cerebellum, optic nerve, cauda equina, hypothalamus, and brain stem.[3]
The most defining physical symptom of astroblastoma, regardless of location, is elevated intracranial pressure, occurring when cerebrospinal fluid in the subarachnoid space exhibits heavy pressure and decreased blood flow, resulting in throbbing headache or nausea for the patient.[2] Despite widespread localization in the brain, astroblastoma is rarely reported in oncological studies, accounting for only 0.45–2.8% of all brain gliomas since its discovery in 1926.[4][5][6] Without a doubt, astroblastoma remains one of the most challenging and problematic tumors to diagnose and treat among all nervous system cancers.
Classification
The World Health Organization, a specialized agency that classifies abnormal tumors affecting the central nervous system and assesses potential risk to life, has difficulty in assigning a proper grade for astroblastoma.[2] The organization’s most recent grade in 2007 assigned astroblastoma as a high-grade III and grade IV neoplasm, signifying that the glial tumor is dangerous for patients, causing fatal problems even after surgery.[5][6] However, recent data compilation from 2011, one that compiled nearly 30 years of clinical information, confirms opposite results from patients: a 95% survival rate exists after astroblastoma is completely removed (gross total resection).[2][4][7] The most important factor for any patient when cancer is concerned – the likelihood of surviving – is still controversial for astroblastoma, but recent advances in the last decade have improved prognosis.
Subtypes
Astroblastoma can be divided into low-grade, well-differentiated tumors and high-grade, anaplastic subtypes.[1] The majority of tumors exhibit a spherical perimeter with either a solid or cystic interior, comprising peripheral vasculature and epithelioid neoplasms.
General Pathology
Since the early 1890s, astroblastoma has established a stable set of pathological qualities that truly distinguishes itself as a separate and significant entity. Compilations from various case reports reveal the following common characteristics:[2]
- Appears "bubbly" in nature
- Polarized, unipolar in structure
- Peripheral vasculature
- Radial arrangement as a pseudorosette
- Immunoreactive for GFAP and vimentin (supports astrocytic origin)
- Lacks "true rosette" architecture
- Lacks structural cohesiveness
- Prominent pseudopapillae formation
- Localization mostly in cerebral hemispheres
- Nodular, non-invasive growth
- Strong, associative vasculature in other parts of the body
- Tissue fibrosis prominence
- High likelihood of vascular hyalinization
- Low likelihood to metastasize toward other regions of the brain
Abnormal Pathology
Beyond normal pathologies, scientists have discovered some abnormal characteristics of astroblastoma in a variety of patients. The presence of a bulky calcification with punctate (pointed) and globular features was noted in a 2009 study of a 12-year-old girl.[8] Computerized tomography confirmed these calcified masses in the posteroinferior region to the fourth ventricle just above the midline. The mass began at the brainstem, extended along the inferior cerebellar peduncle to roof areas against the ventricles through the nodule of vermis, easily detected against normal grey matter surrounding it.[8] Calcification deriving from nervous system tumors is a rare quality in astroblastoma patients, but it is nonetheless easy to identify. Lumbar pain and lower body weakness is also a rarity in astroblastoma patients, even though it is entirely possible for lesions to proliferate toward the spinal cord.[6]
Associations with Other Tumors
An enormous difficulty lies in classifying an astroblastoma tumor due to its overlapping features with other brain tumors. Certain neuroradiologic features finally distinguish astroblastoma from the common ependymoma, another frequent tumor occurring in the fourth ventricle. In general, when brain lesions are smaller than Grade I, demarcating between these features is near impossible, often mistaking astroblastoma with glial neoplasms, high-grade astrocytes, and embryonal neoplasms. However, the “bubbly” appearance in astroblastoma is entirely exclusive.
Researchers have also confirmed astroblastoma distinct from oligodendroglioma, which are invasive nodular cysts that may resemble a "bubbly" interior. Pleomorphic xanthoastrocytoma, dysembryoplastic neuroepithelial tumors, juvenile pilocytic astrocytoma, and hemangioblastoma are well-established, pediatric brain cancer tumors that are often confused with astroblastoma patients. However, further histology has confirmed that special structures and characteristics are unique to astroblastoma. Advances in the 21st century of histology have justified proper diagnosis, eliminating inconsistency that plagued this tumor for several decades.
Research going back to early 2000 marks the first complications for satisfying requirements in radiographic and histopathologic studies. Seven astroblastoma cases of comparative genomic hybridization, a molecular technique analyzing chromosomal changes in DNA content of brain cancer cells, suggested that chromosome 19 and chromosome 20q were amplified in astroblastoma cells throughout the brain.[5][6] These genomic features are responsible for widespread proliferation, tumorigenesis, and deregulation of pathways associated with normal housekeeping. Furthermore, the absence of chromosome function in 9q, 10, and X were not observed in other types of neoplasms, such as an ependymoma.
Specific neuronal markers further distinguish astroblastoma. Neuron-specific enolase (NSE) positive, NSE negative, synaptophysin negative, neurofilament negative, TUJ1 positive, and nestin positive have been expressed in astroblastoma cell populations, showing significant promise in neuronal stem cell treatment for the tumor.[6]
Clinical Symptoms
The majority of patients with astroblastoma display a limited set of physical and physiological symptoms. Rare cases in literature reveal atypical conditions, but these are often exclusive to the individual and do not suggest a widespread trend. As research continues, a larger set of symptoms can be properly assessed in the clinic.
Intracranial pressure
Most patients experience a series of intermittent headaches over a few weeks or sustained, powerful pressure in a matter of days.[2][6] The time-frame for this pressure varies from patient to patient and fluctuate based on the stage of the tumor. Both low-grade and high-grade astroblastoma can exhibit significant discomfort from headaches, although literature supports that higher-grade astroblastoma affect a patient with day-to-day activities, forcing individuals to stay at home away from their jobs and family. Malignant astroblastoma distorts the function of surrounding brain regions, and pressure is the primary result.[1][8][9]
Enhanced drowsiness
Along with cranial pressure, patients exhibit noticeable lethargy, increasing in severity as the tumor progresses.[9][10][11] In the first few months, morning activities are usually unaffected; over time, these effects become more pronounced, especially late at night.[2][6][8] Lethargy can disrupt vital signs, depleting energy and desire to perform simple cognitive tasks.
Frequent nausea
The desire to eat normally becomes worse over time, leading to weight loss from vomiting.[2][10][12] Nausea is seen in almost all cases of astroblastoma, especially in low-grade tumors.
Impaired vision
Vision deficit usually occurs when lesions grow in the occipital lobe of the brain,[1] causing a blurred daze for patients, especially in sensitivity to light. Focusing upon finer objects becomes a challenge, along with edge and border detection.[6][12] Driving behind the wheel is dangerous when astroblastoma grows in residual tissue size, since peripheral vision can be insufficient. Horizontal nystagmus and other involuntary eye disorders can occur.[2][10]
Motor system imbalance and weakness
Frequent reports show that adolescents and adults with grade III and IV astroblastoma fall frequently before they even reach a doctor's office.[11] Alertness is diminished when walking normally, forcing patients to exhibit awkward gait patterns to avoid imbalance.[6][12]
Decreased sensation
Since the motor system can be impaired with severe cases, the malignant spread of astroblastoma throughout the body may press against or paralyze the spinal cord, diminishing sensation in upper and lower extremities.[6][9]
Seizures
Convulsions are observed in older patients with astroblastoma.[9][12]
Psychotic episodes
Grade III and IV astroblastoma have been shown gradually change the mental stability of a patient.[2] Hallucinations impair cognition to the point where patients a lost of identity, although this is not commonly seen in clinic.[6]
Cognitive dysregulation
Irritability, aggression, memory loss, neurological deficits, and inattentiveness on everyday tasks are the most common forms of deregulation in the mental capabilities of a patient.[11] Verbal communication is affected, but usually not to the point where close friends can detect that the individual is cognitively impaired.[8][10]
Epidemiology
Astroblastoma predominantly affects children, but young adults are also susceptible to the tumor .[2] Although the tumor is widely considered a pediatric disease, elderly patients are documented throughout literature.[4][12]
Age incidence
The age distribution of astroblastoma is largely bimodal, suggesting that two distinct diagnosis peaks occur from ages 5–10 and ages 21–30.[2][7] A likely explanation for this discrepancy is that parents of children are more likely to report symptoms of nausea and constant headaches than young adults, who may, at first, disregard these symptoms for a lesser condition. Nevertheless, a combination of age, anatomic location, and image assessment can efficiently evaluate astroblastoma. Furthermore, the age of a patient can aid an oncologist in recommending appropriate treatment plans, along with other factors.
Gender incidence
In reported cases of the tumor over the last 25 years, the number of affected females with astroblastoma is significantly higher than the number of affected males.[1][13] Sughrue et al. confirmed this trend, stating that 70% of the cases with clearly stated gender were female (100 cases total).[2] While several publications support a genetic predisposition to females, the underlying reasons are still unknown.[4][12]
Environmental incidence
At this point, no literature has indicated whether environmental factors increase the likelihood of astroblastoma. Although cancer in general is caused by a variety of external factors, including carcinogens, dangerous chemicals, and viral infections, astroblastoma research has not even attempted to classify incidence in this regard. The next few decades will aid in this understanding.
Recurrence
More than other brain tumors, astroblastoma is frequently a recurring tumor; its rate remains high, even after resection as treatment. Currently, an unfavorable prognosis exists for patients with high-grade, anaplastic astroblastoma: they tend to recur almost indefinitely, forcing the patient to invest in more invasive surgeries. In contrast, a favorable prognosis exists for patients with well-differentiated, low-grade astroblastoma, since patients usually never require such a treatment[9][14][15] The strict black-and-white diagnosis of an astroblastoma based on grade does not determine all tumor behaviors, but it can be used as a benchmark for patients with varying degrees of severity.
Low-grade
The likelihood of low-grade astroblastoma returning after surgery is highly improbable, but some patients have exhibited recurrence.[11][15] Patients with low-grade lesions can remain asymptomatic after surgery and show recurrence 1–2 years in follow-up sessions.[7] However, since residual tissue size is a large determinant for profiling recurrence, it is almost never the case that a low-grade astroblastoma continues to appear in size and strength after the second resection.[14] Usually, patients are not recommended for resection at all and are simply directed towards other therapeutic techniques. Most children can continue to lead productive, healthy lives after a low-grade astroblastoma is treated.
High-grade
Surviving the symptoms of high-grade astroblastoma is not life-threatening, but a significant portion of patients die due to repeated recurrence of tumors as they continue to grow and spread. Unlike conventional low-grade tumors, high-grade tumors associate a plethora of factors when they metastasize to other areas of the body. Therefore, complications frequently occur after surgery is performed since an oncologist cannot efficiently control the tumor in a suitable time-frame.[13] Cases in literature confirm that high-grade patients face up to five or six resection surgeries and still experience symptoms post-operatively.[9][14] The dual-action of chemotherapy and radiotherapy can slow down recurrence when gross total resection is performed multiple times, but there is no guarantee that the tumor will ever be in remission.
Treatment
Like most tumors in the brain, astroblastoma can be treated through surgery and various forms of therapy. Many publications within the last decade have suggested a noticeable improvement in success rate of patients. With the advancement of cutting-edge technology and novel approaches in stem cells, patients are hopeful that they be happy and healthy through old age.
The following factors influence an oncologist's specific treatment plan:
- Patient's overall medical history
- Localization and grade severity of the tumor
- Age and tolerance to certain medications, procedures, and treatment
- Predicted progress of recovery
- Final anticipated outcome of treatment
Gross-total resection
Complete surgical removal, known as gross-total resection or craniotomy, remains the standard for treating astroblastoma, despite high recurrence rate for high-grade tumors.[13] Since there are so few cases reported around the world each year, the standard for surgery varies from physician to physician and is often difficult to rightfully diagnose. Low-grade astroblastomas exhibit low recurrence rates following resection, but varying reports prove that some patients, despite the severity of the lesion, will unpredictably witness recurrence.[9] In a recent study of a 17-year-old male, a low-grade astroblastoma was resected and recurred within 5 months of the therapy, forcing the oncologist to administer further chemotherapy, radiotherapy, and a second resection to completely put the tumor in remission.[15]
Radiotherapy
Radiation therapy selectively kills astroblastoma cells while leaving surrounding normal brain tissue unharmed. The use of radiation therapy after an astroblastoma excision has variable results.[2] Conventional external beam radiation has both positive and negative effects on patients, but it is not recommended at this point to treat all types.[2] All in all, the radiosensitivity of astroblastoma to therapy remains unclear, since some research advocate its effectiveness while others diminish the effects. Future studies must be done on patients with both total excision and sub-excision of the tumor to accurately assess whether radiation benefits patients under different circumstances.
Chemotherapy
Chemotherapy is the preferred secondary treatment after resection. The treatment kills astroblastoma cells left behind after surgery and induces a non-dividing, benign state for remaining tumor cells. Normally, chemotherapy is not recommended until the second required resection, implying that the astroblastoma is a high-grade tumor continuing to recur every few months.[1][13] A standard chemotherapy protocol starts with two rounds of nimustine hydrochoride (ACNU), etoposide, vincristine, and interferon-beta.[6] The patient undergoes a strict drug regimen until another surgery is required. By the third surgery, should recurrence in the astroblastoma occur, a six-round program of ifosfamide, cisplatin, and etoposide will "shock" the patient's system to the point where recurrence halts.[6] Unfortunately, chemotherapy may not always be successful with patients requiring further resection of the tumor, since the tumor cell begins to show superior vasculature and a strong likelihood of compromising a patient's well-being. Oral ingestion of temozolomide for at-home bedside use may be preferred by the patient.
Future advances
A popular form of surgery involves CyberKnife radiotherapy and Gamma Knife radiosurgery. Their success-rate on cranial lesion is fairly effective, but recurrence is still a problem for severe patients.[6]
One of the more exciting and promising routes for treatment involves stem cell use to combat astroblastoma. A study in 2005 profiled cell surface markers of astroblastoma cells removed from an 11-year-old patient. Fluorescence activation suggested that about 1/4 of these cells were CD133 positive CD24, CD34, and CD45 negative. This specific genetic makeup lends to self-renewal, differentiation, and propagation of neural stem cells in the brain.[16] However, the work remains a preliminary insight into the role of neuronal stemlike cells on astroblastoma development.[16] Gene therapy and immunotherapy are also possible considerations for specialized astroblastoma therapies, but they are not mentioned in current literature.
See also
References
- 1 2 3 4 5 6 Unal, Ekrem, and Yavuz Koksal. "Astroblastoma in a Child." Children Nervous System 24.2 (2008): 165–68.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Sughrue, Michael E., Jay Choi, Martin Rutkowski, and Derick Aranda. "Clinical Features and Post-surgical Outcome of Patients with Astroblastoma." Journal of Clinical Neuroscience 18.6 (2011): 750–54.
- ↑ Denaro, Luca, Marina Gardiman, and Milena Caliderone. "Intraventricular Astroblastoma. Case Report." Journal of Neurosurgery Pediatrics 1 (2008): 152–55.
- 1 2 3 4 Bell, John W., Anne G. Osborn, Karen L. Salzman, Susan I. Blaser, Blaise V. Jones, and Steven S. Chin. "Neuroradiologic Characteristics of Astroblastoma." Diagnostic Neuroradiology 49 (2007): 203–09.
- 1 2 3 Brat, Daniel J., Yuichi Hirose, Kenneth J. Cohen, Burt G. Feuerstein, and Peter C. Burger. "Astroblastoma: Clinicopathologic Features and Chromosomal Abnormalities Defined by Comparative Genomic Hybridization." Brain Pathology 10.3 (2000): 342–52.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Hirano, Hirofumi, Shunji Yunoue, Masatomo Kaji, Masahiro Tsuchiya, and Kazunori Arita. "Consecutive Histological Changes in an Astroblastoma That Disseminated to the Spinal Cord after Repeated Intracranial Recurrences: a Case Report." Brain Tumor Pathology 25.1 (2008): 25–31.
- 1 2 3 Lau, Patrick, Teresa Thomas, Philip Lui, and Aye Khin. "‘Low‐grade’ Astroblastoma with Rapid Recurrence: a Case Report." Pathology 38.1 (2006): 78–80.
- 1 2 3 4 5 Ganapathy, Srinivas, Laurence I. Kleiner, David L. Mirkin, and Emmett Broxson. "Unusual Manifestations of Astroblastoma: a Radiologic–pathologic Analysis." Pediatric Radiology 39.2 (2009): 168–71.
- 1 2 3 4 5 6 7 Bonnin JM, Rubinstein LJ. "Astroblastoma: a pathological study of 23 tumors, with a postoperative follow-up in 13 patients." Neurosurgery 25.1 (1989): 6–13.
- 1 2 3 4 Kubota, Toshihiko, Kazufumi Sato, Hidetaka Arishima, Hiroaki Takeuchi, Ryuhei Kitai, and Takao Nakagawa. "Astroblastoma: Immunohistochemical and Ultrastructural Study of Distinctive Epithelial and Probable Tanycytic Differentiation." Neuropathology 26.1 (2006): 72–81.
- 1 2 3 4 Kemerdere, Rahsan, Reza Dashti, and Mustafa Ulu. "Supratentorial High Grade Astroblastoma: Report of Two Cases and Review of the Literature." Turkish Neurosurgery 19.2 (2009): 149–52.
- 1 2 3 4 Weintraub, David, Stephen Monteith, and Chun Po Yen. "Recurrent Astroblastoma Treated with Gamma Knife Radiosurgery." Journal of Neurooncology 103.3 (2011): 751–54.
- 1 2 3 W. Mierau, R. Weslie Tyson, Loris M, Gary. "Astroblastoma: Ultrastructural Observations on a Case of High-Grade Type." Ultrastructural Pathology 23.5 (1999): 325–32.
- 1 2 3 Masamoto, Kaji, and Takeshima Hideo. "Low-Grade Astroblastoma Recurring With Extensive Invasion-Case Report." (2006).
- 1 2 Huhn, Stephen L., Yun Yung, Samuel Cheshier, Griffith Harsh, Laurie Ailles, Irving Weissman, Hannes Vogel, and Victor Tse. "Identification of Phenotypic Neural Stem Cells in a Pediatric Astroblastoma." Journal of Neurosurgery: Pediatrics 103.5 (2005): 446–50.