Blau syndrome
| Blau syndrome | |
|---|---|
|
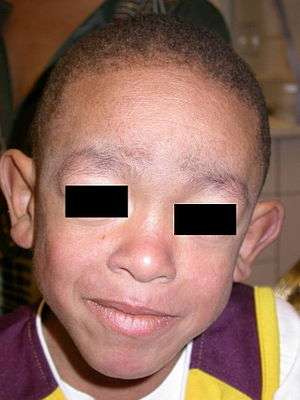
Coarse facial features in a boy with Blau syndrome | |
| Classification and external resources | |
| OMIM | 186580 |
| DiseasesDB | 32725 |
In 1985 Edward Blau, a pediatrician in Marshfield Wisconsin, reported a family that over four generations had granulomatous inflammation of the skin, eyes and joints. The condition was transmitted as an autosomal dominant trait. In the same year Jabs et al. reported a family that over two generations had granulomatous synovitis, uveitis and cranial neuropathies. The condition was transmitted in an autosomal dominant fashion. In 1981 Malleson et al. reported a family that had autosomal dominant synovitis, camptodactyly, and iridocyclitis. One member died of granulomatous arteritis of the heart and aorta. In 1982 Rotenstein reported a family with granulomatous arteritis, rash, iritis, and arthritis transmitted as an autosomal dominant trait over three generations. Then in 1990 Pastores et al. reported a kindred with a phenotype very similar to what Blau described and suggested that the condition be called Blau Syndrome (BS). They also pointed out the similarities in the families noted above to BS but also pointed out the significant differences in the phenotypes. In 1996 Tromp et al. conducted a genome wide search using affected and non affected members of the original family. A marker D16S298gave a maximum LOD score of 3.75 and put the BS susceptibility locus within the 16p12-q21 interval. Hugot et al. found a susceptibility locus for Crohn disease a granulomatous inflammation of the bowel on chromosome 16 close to the locus for BS. Based on the above information Blau suggested in 1998 that the genetic defect in BS and Crohn Disease might be the same or similar.
Finally in 2001 Miceli-Richard et al. found the defect in BS to be in the nucleotide-binding domain of CARD15/NOD2. They commented in their article that mutations in CARD15 had also been found in Crohn Disease. Confirmation of the defect in BS being in the CARD15 gene was made by Wang et al. in 2002 using the BS family and others.(
With that information the diagnosis of BS was not only determined by phenotype but now by genotype.
Early onset sarcoidosis is BS without a family history, BS has been diagnosed in patients who have not only the classic triad but granuloma in multiple organs. Treatment has included the usual anti inflammatory drugs such as adrenal glucocorticoids, anti metabolites and also biological agents such as anti-TNF and infliximab all with varying degrees of success.
The elucidation that the gene defect in BS involves the CARD15/NOD2 gene has stimulated many investigators to define how this gene operates as part of the innate immune system that responds to bacterial polysaccharides such as muramyl dipeptide to induce signaling pathways that induce cytokine responses that protect the organism. In BS the genetic defect seems to lead to over expression and poor control of the inflammatory response leading to widespread granulomatous inflammation and tissue damage. This reference provides an excellent review of not only the clinical aspects of BS but also the presumed pathogenetic mechanisms brought about by the gene defect.
Unanswered questions are what is the stimulus that activates the aberrant immune response and if found could more precise therapy be used and what is the relationship to the specific gene defect and the phenotype?
External links
References
- Wouters CH, Maes A, Foley KP et al. Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J 2014;12:33 doi:10.1186/1546-0096-12-33 [PMC free article] [PubMed]
- Blau EB : Familial Granulomatous Arthritis, Iritis and Rash. J Pediatr 1985 ;107: 689-693
- Jabs DA, Houk JL, Bias WB and Arnett FC: Familial Granulomatous Synovitis, Uveitis, and Cranial Neuropathies. Am J Med 1985; 78: 801-804.
- Malleson P, Schaller JG, Dega F, Cassidy SB andPagon RA : Familial Arthritis and Camplodactyly. Arthtitis Rheum 1981; 24: 1199-1203.
- Rotenstein D, Gibbas DL, Mujmudar B, and Chastain E: Familial Granulomatous Arteritis with Polyarthritis of Juvenile Onset. N Engl J Med 1982; 306: 85-90.
- Pastores GM, Michels VV, Stickler GB et al. : Autosomal dominant granulomatous arthritis, uveitis, skin rash, and synovial cysts. J Pediatr 1990; 117: 403-408.
- Tromp G, Kuivaniemi H, Raphael S, et al.: Genetic Linkage of Familial Granulomatous Inflammatory Arthritis, Skin Rash, and Uveitis to Chromsome 16. Am J Hum Genet 1996;59:1097-1107.
- Hugot JP: Mutations in CARD15 associated with Crohn Disease. Nature 2001; 411:599-603.
- Blau EB : Autosomal dominant granulomatous disease of childhood: The naming of things. J Pediatr 1998; 133: 322-3.
- Miceli-Richard C, Lesage S, Rybojad M et al. : CARD!% mutations in Blau syndrome. Nature Genetics 2005; 29: 1920.
- Wang X, Kuivaniemi H, Bonvita G et al.: CARD 15 Mutations in Familial Granulomatous Syndromes. Arthritis Rheum 2002; 46:3041-3045.
- Caso F, Galozzi P, Costa L et al.; Autoinflammatory granulomatous disease: from Blau syndrome and early-onset sarcoidosis to NOD2-mediated diseased Crohn's disease. RMD Open 2015; 1:e000097.
