Knorr pyrrole synthesis
| Knorr pyrrole synthesis | |
|---|---|
| Named after | Ludwig Knorr |
| Reaction type | Ring forming reaction |
| Identifiers | |
| RSC ontology ID | RXNO:0000497 |
The Knorr pyrrole synthesis is a widely used chemical reaction that synthesizes substituted pyrroles (3).[1][2][3] The method involves the reaction of an α-amino-ketone (1) and a compound containing a methylene group α- to (bonded to the next carbon to) a carbonyl group (2).[4]
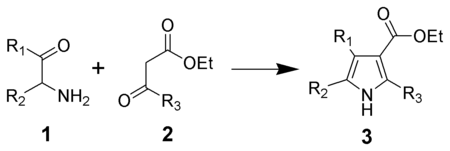
Method
The mechanism requires zinc and acetic acid as catalysts. It will proceed at room temperature. Because α-amino-ketones self-condense very easily, they must be prepared in situ. The usual way of doing this is from the relevant oxime.[5][6]
The original Knorr synthesis employed two equivalents of ethyl acetoacetate, one of which was converted to ethyl 2-oximinoacetoacetate by dissolving it in glacial acetic acid, and slowly adding one equivalent of saturated aqueous sodium nitrite, under external cooling. Zinc dust was then stirred in, reducing the oxime group to the amine. This reduction consumes two equivalents of zinc and four equivalents of acetic acid.
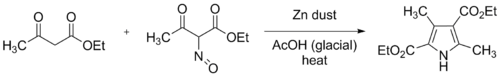
Modern practice is to add the oxime solution resulting from the nitrosation and the zinc dust gradually to a well-stirred solution of ethyl acetoacetate in glacial acetic acid. The reaction is exothermic, and the mixture can reach the boiling point, if external cooling is not applied. The resulting product, diethyl 3,5-dimethylpyrrole-2,4-dicarboxylate, has been called Knorr's Pyrrole ever since. In the Scheme above, R2 = COOEt, and R1 = R3 = Me represent this original reaction.
Knorr's pyrrole can be derivatized in a number of useful manners. One equivalent of sodium hydroxide will saponify the 2-ester selectively. Dissolving Knorr's pyrrole in concentrated sulfuric acid, and then pouring the resulting solution into water will hydrolyze the 4-ester group selectively. The 5-methyl group can be variously oxidized to chloromethyl, aldehyde, or carboxylic acid functionality by the use of stoichiometric sulfuryl chloride in glacial acetic acid.[7] Alternatively, the nitrogen atom can be alkylated. The two ester positions can be more smoothly differentiated by incorporating benzyl or tertiary-butyl groups via the corresponding acetoacetate esters. Benzyl groups can be removed by catalytic hydrogenolysis over palladium on carbon, and tertiary-butyl groups can be removed by treatment with trifluoroacetic acid, or boiling aqueous acetic acid. R1 and R3 (as well as R2 and "Et") can be varied by the application of appropriate beta-ketoesters readily made by a synthesis emanating from acid chlorides, Meldrum's acid, and the alcohol of one's choice. Ethyl and benzyl esters are easily made thereby, and the reaction is noteworthy in that even the highly hindered tertiary-butyl alcohol gives very high yields in this synthesis.[8]
Levi and Zanetti extended the Knorr synthesis in 1894 to the use of acetylacetone (2,4-pentanedione) in reaction with ethyl 2-oximinoacetoacetate. The result was ethyl 4-acetyl-3,5-dimethylpyrrole-2-carboxylate, where "OEt" = R1 = R3 = Me, and R2 = COOEt.[9] The 4-acetyl group could easily be reduced to a 4-ethyl group by use of the Wolff-Kishner reduction (hydrazine and alkali, heated); hydrogenolysis, or the use of diborane. Benzyl or tertiary-butyl acetoacetates also work well in this system, and with close temperature control, the tertiary-butyl system gives a very high yield (close to 80%).[10] N,N-dialkyl pyrrole-2- and/or 4-carboxamides may be prepared by the use of N,N-dialkyl acetoacetamides in the synthesis. Even thioesters have been successfully prepared, using the method.[11] As for the nitrosation of beta-ketoesters, despite the numerous literature specifications of tight temperature control on the nitrosation, the reaction behaves almost like a titration, and the mixture can be allowed to reach even 40 °C without significantly impacting the final yield.
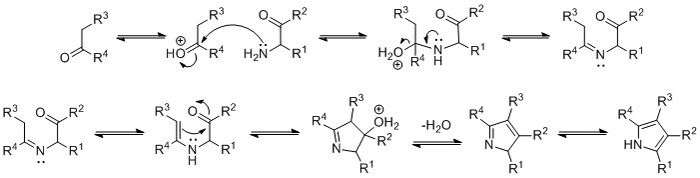
The mechanism of the Knorr pyrrole synthesis begins with condensation of the amine and ketone to give an imine. The imine then tautomerizes to an enamine, followed by cyclization, elimination of water, and isomerization to the pyrrole.
Related synthesis
There are a number of important syntheses of pyrroles that are operated in the manner of the Knorr Synthesis, despite having mechanisms of very different connectivity between the starting materials and the pyrrolic product.
Fischer and Fink found that Zanetti's synthesis from 2,4-pentanedione and ethyl 2-oximinoacetoacetate gave ethyl 3,5-dimethylpyrrole-2-carboxylate as a trace byproduct. Similarly, 3-ketobutyraldehyde diethyl acetal led to the formation of ethyl 5-methylpyrrole-2-carboxylate. Both of these products resulted from the loss of the acetyl group from the inferred ethyl 2-aminoacetoacetate intermediate. An important product of the Fischer-Fink synthesis was ethyl 4,5-dimethylpyrrole-2-carboxylate, made from ethyl 2-oximinoacetoacetate and 2-methyl-3-oxobutanal, in turn made by the Claisen condensation of 2-butanone with ethyl formate.[12]
G.G. Kleinspehn reported that the Fischer–Fink connectivity could be forced to occur exclusively, by the use of diethyl oximinomalonate in the synthesis, with 2,4-pentanedione, or its 3-alkyl substituted derivatives. Yields were high, around 60%, and this synthesis eventually came to be one of the most important in the repertory.[13] Yields were significantly improved, by the use of preformed diethyl aminomalonate (prepared by the hydrogenolysis of diethyl oximinomalonate in ethanol, over Pd/C), and adding a mixture of diethyl aminomalonate and the beta-diketone to actively boiling glacial acetic acid.[14]
Meanwhile, Johnson had extended the Fischer-Fink synthesis by reacting 2-oximinoacetoacetate esters (ethyl, benzyl, or tertiary-butyl), with 3-alkyl substituted 2,4-pentanediones.[15] Others extended the Kleinspehn synthesis by the use of unsymmetrical beta-diketones (such as 3-alkyl substituted 2,4-hexanediones), which preferentially reacted initially at the less hindered acetyl group and afforded the corresponding 5-methylpyrrole-2-carboxylate esters. N,N-Dialkyl 2-oximinoacetoacetamides also were found to give pyrroles when reacted under Knorr conditions with 3-substituted-2,4-pentanediones, in yields comparable to the corresponding esters (around 45%). However, when unsymmetrical diketones were used, it was found that the acetyl group from the acetoacetamide was retained in the product, and one of the acyl groups from the diketone had been lost.[16] This same mechanism occurs to a minor extent in the acetoacetate ester systems, and had previously been detected radiochemically by Harbuck and Rapoport.[17] Most of the above-described syntheses have application in the synthesis of porphyrins, bile pigments, and dipyrrins.
References
- ↑ Knorr, L. (1884). "Synthese von Pyrrolderivaten" [Synthesis of pyrrole derivatives]. Berichte der deutschen chemischen Gesellschaft. 17 (2): 1635–1642. doi:10.1002/cber.18840170220.
- ↑ Knorr, L. (1886). "Synthetische Versuche mit dem Acetessigester" [Synthetic experiments with the ester of acetoacetic acid]. Annalen der Chemie. 236: 290–332. doi:10.1002/jlac.18862360303.
- ↑ Knorr, L.; Lange, H. (1902). "Ueber die Bildung von Pyrrolderivaten aus Isonitrosoketonen" [On the formation of pyrrole derivatives from isonitrosoketones]. Berichte der deutschen chemischen Gesellschaft. 35 (3): 2998–3008. doi:10.1002/cber.19020350392.
- ↑ Corwin, Alsoph Henry, "Chapter 6: The Chemistry of Pyrrole and its Derivatives," in: Robert Cooley Elderfield, ed., Heterocyclic Compounds, volume 1 (New York, New York: Wiley, 1950), pp. 287 ff.
- ↑ Fischer, H. Organic Syntheses, Coll. Vol. 2, p. 202 (1943); Vol. 15, p. 17 (1935). (Article)
- ↑ Fischer, H. Organic Syntheses, Coll. Vol. 3, p. 513 (1955); Vol. 21, p. 67 (1941). (Article)
- ↑ Corwin, A. H.; Bailey, W. A.; Viohl, P. (1942). "Structural Investigations upon a Substituted Dipyrrylmethane. An Unusual Melting Point-Symmetry Relationship1,2". Journal of the American Chemical Society. 64 (6): 1267. doi:10.1021/ja01258a007.
- ↑ Oikawa, Y.; Sugano, K.; Yonemitsu, O. (1978). "Meldrum's acid in organic synthesis. 2. A general and versatile synthesis of .beta.-keto esters". The Journal of Organic Chemistry. 43 (10): 2087. doi:10.1021/jo00404a066.
- ↑ Zanetti, C.U.; Levi, E. (1894). "Sintesi di composti pirrolici dai nitrosochetoni" [Synthesis of pyrrole compounds from nitrosoketones]. La Gazzetta Chimica Italiana. 24 (1): 546–554.
- ↑ Treibs, A.; Hintermeier, K. (1954). "Tert.-Butylester von Pyrrolcarbonsäuren". Chemische Berichte. 87 (8): 1167. doi:10.1002/cber.19540870818.
- ↑ Bullock, E.; Chen, T.S.; Loader, C.E. (1966). "Preparation and reactions of some pyrrylthiol esters". Canadian Journal of Chemistry. 44 (9): 1007–11. doi:10.1139/v66-149.
- ↑ Fischer, Hans; Fink, Emmy (1948). "Über eine neue Pyrrolsynthese" [On a new synthesis of pyrroles]. Zeitschrift für Physiologische Chemie. 283: 152–161. doi:10.1515/bchm2.1948.283.3-4.152.
- ↑ Kleinspehn, G. G. (1955). "A Novel Route to Certain 2-Pyrrolecarboxylic Esters and Nitriles1,2". Journal of the American Chemical Society. 77 (6): 1546. doi:10.1021/ja01611a043.
- ↑ Paine, J. B.; Dolphin, D. (1985). "Pyrrole chemistry. An improved synthesis of ethyl pyrrole-2-carboxylate esters from diethyl aminomalonate". The Journal of Organic Chemistry. 50 (26): 5598. doi:10.1021/jo00350a033.
- ↑ Bullock, E.; Johnson, A. W.; Markham, E.; Shaw, K. B. (1958). "287. A synthesis of coproporphyrin III". Journal of the Chemical Society (Resumed): 1430. doi:10.1039/JR9580001430.
- ↑ Paine, J. B.; Brough, J. R.; Buller, K. K.; Erikson, E. E.; Dolphin, D. (1987). "Mechanism of the formation of N,N-dialkyl-2-pyrrolecarboxamides from 1,3-diketones and N,N-dialkyloximinoacetoacetamides". The Journal of Organic Chemistry. 52 (18): 3993. doi:10.1021/jo00227a010.
- ↑ Rapoport, H.; Harbuct, J. W. (1971). "Mechanism of a modified Knorr pyrrole condensation". The Journal of Organic Chemistry. 36 (6): 853. doi:10.1021/jo00805a030.
