MID1
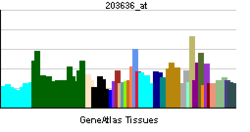
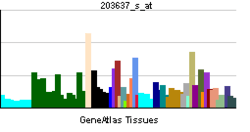
| View/Edit Human | View/Edit Mouse |
Midline-1 is a protein that in humans is encoded by the MID1 gene.[3][4][5]
Function
The protein encoded by this gene is a member of the tripartite motif (TRIM) family, also known as the 'RING-B box-coiled coil' (RBCC) subgroup of RING finger proteins. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. This protein forms homodimers which associate with microtubules in the cytoplasm. The protein is likely involved in the formation of multiprotein structures acting as anchor points to microtubules. Mutations in this gene have been associated with the X-linked form of Opitz syndrome, which is characterized by midline abnormalities such as cleft lip, laryngeal cleft, heart defects, hypospadias, and agenesis of the corpus callosum. This gene was also the first example of a gene subject to X inactivation in human while escaping it in mouse. Several different transcript variants are generated by alternate splicing; however, the full length nature of two variants has not been determined.[5]
Interactions
MID1 has been shown to interact with MID2.[6][7]
References
- ↑ "Human PubMed Reference:".
- ↑ "Mouse PubMed Reference:".
- ↑ Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A (Dec 1997). "Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22". Nat Genet. 17 (3): 285–91. doi:10.1038/ng1197-285. PMID 9354791.
- ↑ Perry J, Feather S, Smith A, Palmer S, Ashworth A (Mar 1998). "The human FXY gene is located within Xp22.3: implications for evolution of the mammalian X chromosome". Hum Mol Genet. 7 (2): 299–305. doi:10.1093/hmg/7.2.299. PMID 9425238.
- 1 2 "Entrez Gene: MID1 midline 1 (Opitz/BBB syndrome)".
- ↑ Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A (May 2001). "The tripartite motif family identifies cell compartments". EMBO J. 20 (9): 2140–51. doi:10.1093/emboj/20.9.2140. PMC 125245
 . PMID 11331580.
. PMID 11331580. - ↑ Short KM, Hopwood B, Yi Z, Cox TC (2002). "MID1 and MID2 homo- and heterodimerise to tether the rapamycin-sensitive PP2A regulatory subunit, alpha 4, to microtubules: implications for the clinical variability of X-linked Opitz GBBB syndrome and other developmental disorders". BMC Cell Biol. 3: 1. doi:10.1186/1471-2121-3-1. PMC 64779. PMID 11806752.
Further reading
- De Falco F, Cainarca S, Andolfi G, Ferrentino R, Berti C, Rodríguez Criado G, Rittinger O, Dennis N, Odent S, Rastogi A, Liebelt J, Chitayat D, Winter R, Jawanda H, Ballabio A, Franco B, Meroni G (2004). "X-linked Opitz syndrome: novel mutations in the MID1 gene and redefinition of the clinical spectrum". Am. J. Med. Genet. A. 120 (2): 222–8. doi:10.1002/ajmg.a.10265. PMID 12833403.
- Robin NH, Feldman GJ, Aronson AL, Mitchell HF, Weksberg R, Leonard CO, Burton BK, Josephson KD, Laxová R, Aleck KA, Allanson JE, Guion-Almeida ML, Martin RA, Leichtman LG, Price RA, Opitz JM, Muenke M (1996). "Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11.2". Nat. Genet. 11 (4): 459–61. doi:10.1038/ng1295-459. PMID 7493033.
- Gaudenz K, Roessler E, Quaderi N, Franco B, Feldman G, Gasser DL, Wittwer B, Horst J, Montini E, Opitz JM, Ballabio A, Muenke M (1998). "Opitz G/BBB syndrome in Xp22: mutations in the MID1 gene cluster in the carboxy-terminal domain". Am. J. Hum. Genet. 63 (3): 703–10. doi:10.1086/302010. PMC 1377398. PMID 9718340.
- Van den Veyver IB, Cormier TA, Jurecic V, Baldini A, Zoghbi HY (1998). "Characterization and physical mapping in human and mouse of a novel RING finger gene in Xp22". Genomics. 51 (2): 251–61. doi:10.1006/geno.1998.5350. PMID 9722948.
- Schweiger S, Foerster J, Lehmann T, Suckow V, Muller YA, Walter G, Davies T, Porter H, van Bokhoven H, Lunt PW, Traub P, Ropers HH (1999). "The Opitz syndrome gene product, MID1, associates with microtubules". Proc. Natl. Acad. Sci. U.S.A. 96 (6): 2794–9. doi:10.1073/pnas.96.6.2794. PMC 15848. PMID 10077590.
- Cainarca S, Messali S, Ballabio A, Meroni G (1999). "Functional characterization of the Opitz syndrome gene product (midin): evidence for homodimerization and association with microtubules throughout the cell cycle". Hum. Mol. Genet. 8 (8): 1387–96. doi:10.1093/hmg/8.8.1387. PMID 10400985.
- Cox TC, Allen LR, Cox LL, Hopwood B, Goodwin B, Haan E, Suthers GK (2000). "New mutations in MID1 provide support for loss of function as the cause of X-linked Opitz syndrome". Hum. Mol. Genet. 9 (17): 2553–62. doi:10.1093/hmg/9.17.2553. PMID 11030761.
- Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A (2001). "The tripartite motif family identifies cell compartments". EMBO J. 20 (9): 2140–51. doi:10.1093/emboj/20.9.2140. PMC 125245. PMID 11331580.
- Liu J, Prickett TD, Elliott E, Meroni G, Brautigan DL (2001). "Phosphorylation and microtubule association of the Opitz syndrome protein mid-1 is regulated by protein phosphatase 2A via binding to the regulatory subunit alpha 4". Proc. Natl. Acad. Sci. U.S.A. 98 (12): 6650–5. doi:10.1073/pnas.111154698. PMC 34408. PMID 11371618.
- Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH, Schneider R, Schweiger S (2001). "MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation". Nat. Genet. 29 (3): 287–94. doi:10.1038/ng762. PMID 11685209.
- Short KM, Hopwood B, Yi Z, Cox TC (2002). "MID1 and MID2 homo- and heterodimerise to tether the rapamycin-sensitive PP2A regulatory subunit, alpha 4, to microtubules: implications for the clinical variability of X-linked Opitz GBBB syndrome and other developmental disorders". BMC Cell Biol. 3: 1. doi:10.1186/1471-2121-3-1. PMC 64779. PMID 11806752.
- Landry JR, Mager DL (2003). "Widely spaced alternative promoters, conserved between human and rodent, control expression of the Opitz syndrome gene MID1". Genomics. 80 (5): 499–508. doi:10.1016/S0888-7543(02)96863-1. PMID 12408967.
- Landry JR, Rouhi A, Medstrand P, Mager DL (2003). "The Opitz syndrome gene Mid1 is transcribed from a human endogenous retroviral promoter". Mol. Biol. Evol. 19 (11): 1934–42. doi:10.1093/oxfordjournals.molbev.a004017. PMID 12411602.
- Winter J, Lehmann T, Suckow V, Kijas Z, Kulozik A, Kalscheuer V, Hamel B, Devriendt K, Opitz J, Lenzner S, Ropers HH, Schweiger S (2003). "Duplication of the MID1 first exon in a patient with Opitz G/BBB syndrome". Hum. Genet. 112 (3): 249–54. doi:10.1007/s00439-002-0901-5. PMID 12545276.
- Granata A, Quaderi NA (2003). "The Opitz syndrome gene MID1 is essential for establishing asymmetric gene expression in Hensen's node". Dev. Biol. 258 (2): 397–405. doi:10.1016/S0012-1606(03)00131-3. PMID 12798296.
- Winter J, Lehmann T, Krauss S, Trockenbacher A, Kijas Z, Foerster J, Suckow V, Yaspo ML, Kulozik A, Kalscheuer V, Schneider R, Schweiger S (2004). "Regulation of the MID1 protein function is fine-tuned by a complex pattern of alternative splicing". Hum. Genet. 114 (6): 541–52. doi:10.1007/s00439-004-1114-x. PMID 15057556.
External links
- Meroni G (2007-06-20). "X-Linked Opitz G/BBB Syndrome". GeneReviews -- NCBI Bookshelf. the University of Washington, Seattle and the National Center for Biotechnology Information.
PDB gallery | ||||
|---|---|---|---|---|
| ||||