Phosphinimide ligands
Phosphinimide ligands, also known as phosphorane iminato ligands, are any of a class of organic compounds of the general formula NPR3−.
Structure and Bonding
Phosphinimide ligands have the general formula NPR3−, where the R groups represent organic substituents or, in rare cases, halides or NR2 groups.[1] A discrete ion of NPR3− is not known, but the ligand can be easily derived from neutral phosphinimine precursors.[1][2]
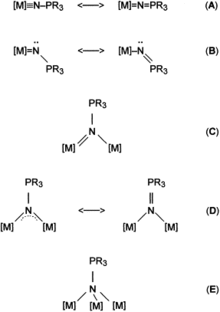
Several coordination types have been observed for transition metal phosphinimide complexes including terminal, μ2-N-bridging, and μ3-N-bridging.[1] In the terminal bonding mode the ligand can have either a linear or a bent geometry at nitrogen, and in the μ2-N-bridging mode, four-membered M2N2 rings can be formed either in an unsymmetrical fashion or in a symmetrical fashion.[1][3] The preferred coordination type is largely dependent upon the oxidation state of the metal as well as, to a lesser extent, upon the ligand sphere of the metal and the steric and electronic properties of the R groups on phosphorus. Bonding modes (A) and (B) are primarily seen in complexes containing metals in high oxidation states, mode (E) is preferred by metals with low oxidation states, and modes (C) and (D) are preferred by metals with medium oxidation states.[1]
In main group phosphinimide complexes, only terminal and μ2-N-bridging bonding modes are observed.[4] The terminally bound ligands are primarily of type (B) with M-N-P bond angles ranging from 120-150°, and often showing surprisingly short M-N bonds.[4] Both the M-N and N-P bond lengths actually come close to being double bonds. This bonding can best be described by a covalent single bond with an overlaying share of polar bonding. The μ2-N-bridging mode arises when the free electron pair at nitrogen gives rise to dimerization.[4]
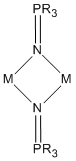
These dimeric complexes yield different M-N bond lengths depending on the ligands present in the rest of the ligand sphere of M.[4] When the complex contains two or four identical ligands, nearly equal M-N distances are observed, whereas, when different or odd-numbered identical ligands are in the complex, the M-N distances are all of significantly different length.[4]
Properties and Reactivity
The phosphinimide ligand is isoelectronic with ligands such as OPR3, [OSiR3]−, [CSiR3]3− and [NSiR3]2−, and often has remarkable similarities in structure and bonding to [M]-OSiR3.[1][4] By varying the R groups on P, a variety of ligands with different electronic and steric properties can be produced, and due to the high oxidation state of phosphorus, these ligands have good thermal stability.[2] Many transition metal phosphinimide complexes have been well-developed and, more recently, main group phosphinimide complexes have been synthesized.[4]
When bound in the terminal linear fashion, the ligand shows steric properties similar to a cyclopentadienyl ligand. However, due to the intervening nitrogen atom, the steric bulk is more removed from the metal center. This property of the ligand can affect the reactivity of the complex by leaving the first coordination sphere of the metal more open, while still having a second coordination sphere environment similar to that of cyclopentadienyl.[2]
Synthesis
Synthesis of Phosphinimines
One of the most useful phosphonimines is R3PNSiMe3 as it is used in the formation of phospinimide complexes. Its formation is quite simple, consisting of the reaction between R3P and R3SiN3, which oxidizes the azide to R3PNSiMe3. The reaction of the lithium phosphinimide salt LiNPR3 with R2SiCl2 also yield R3PNSiMe3. Along with the synthesis of metal complexes, R3PNSiMe3 is an important reagent in the creation R3PNH, as is done through acidic activation with methanol or iso-propanol. R3PNH can also be prepared from salts of the form [R3PNH2]X, which can be prepared through the mixing of PPh3, CCl4, and NH3. Deprotonation of the salt with NaNH2 yields R3PNH.[3][4]
Lithium phosphinimides are also used in the formation of phosphinimide metal complexes. [LiNPR3]4 can be formed through the reaction of R3PNH and LiMe (or LiCl, with possible dimerization).[3][4][5]
Synthesis Phosphinimide Metal Complexes
Group IV and V transition metal phosphinimide complexes have been studied for their catalytic activity in olefin polymerization and, for the most part, determined to be useful catalysts with a high degree of activation. Typical preexisting catalysts involve cyclopentadienyl and chloride substituents on the metal. Therefore, the phosphinimide ligand has typically been substituted into Cp and Cl containing complexes.[3]
Titanium
The synthesis of CpTi(NPR3)Cl2 is achieved with respectable yield through the reaction of R3PNSiMe3 and CpTiCl3. An alternative to the Cp ligand is the tropinyl ligand. If this is preferable, tropinyl can be added through the substitution of Cl in TiCl4, followed by the addition of the phosphinimide through its lithium salt [R3PNHLi]4, diplacing another chloride. Straight addition of the lithium salt to TiCl4 is also possible, displacing up to three chlorides. Alkylation and arylation of the phosphinimide complexes is possible through alkyllithium or Grignard reagents, giving products such as CpTi(NPR3)Me2. Sulfur substituents such as SR can be substituted for the chlorides through addition of LiSR or for the methyls if HSR is added. Several alkyl metallocycle complexes can be achieved through reduction of the metal with Mg along with addition of the appropriate organic molecule (or CO).[3]
Zirconium
Similar zirconium complexes can be formed; however, substitution of the phosphinimide must be done through the addition of the lithium salt, as the R3PNSiMe3 method isn’t effective. Sodium cyclopentadienide can be added to a dihalide phosphinimide to add another Cp or a different Cp ring to the metal and displace a halide. Unlike the titanium complex, the addition of two phosphinimide ligands requires a slightly different route. R3PNH reacted with Zr(NEt2)4, followed by substitution of the remaining NEt2 ligands with Cl through the addition of Me3SiCl yields the bis(phsphinimide) (R3PN)2ZrCl2. Comparable to the titanium complexes, the zirconium complexes can be alkylated or arylated through simple substitution. Once alkylated with Me, reaction with THF and B(C6F5)3 produces the cationic complex [R3PNZr(Cp)Cl(THF)]+.[3]
Vanadium
The methods used for titanium and zirconium aren’t effective with vanadium complexes. Instead of reaction with CpMCl3 or MCl4, R3PNSiMe3 must be reacted with ArNVCl3. Once again, alkylation and arylation are possible, along with substitution of Cl with other ligands such as SCH2Ph and N(SiMe3)2. Although vanadium phosphinimides are isolable, they are not very highly active catalysts.[3]
Tantalum
Phosphinimide tantalum complexes are producible through the straightforward method of adding R3PNSiMe3 to Cp and Cl coordinated Ta complexes. The resulting R3PNTa(Cp)Cl3 complex can be alkylidated with Me or CH2Ph. In the addition of CH2Ph through its Grignard reagent, an alkylidene complex is formed, steming from one CH2Ph adding as the alkylidene and the other coordinating as a single bond. If MeI is introduced, the alkylidene is transformed to a metallocycle, with the Me bonding to the metal coordinated carbon and the metal. Similarly, the addition of EtMgCl forms a metallocycle, with the second equivalent displacing the Cl and coordinating as Et. Finally, cationic complexes can also be formed, generally through the addition of B(C6F5)4. If added to the alkylidated Ta phosphinimide with Ph3C, a methyl group is dissociated; however, if added to the metallocycle, the Ph3C will bond to one of the carbons of the metallocycle, breaking its bond with the metal. If the Ph3C is left out and only B(C6F5)3 is used instead, the boron atom will open the ring by bonding to one of the metal, leaving a negative charge on the boron and a positive charge on the metal.[3]
Other Metals
Other metals such as tin and antimony react with phosphinimide ligands in a similar matter, through chlorinated complexes with R3PNSiMe3.[4]
Applications
Phosphinimide Polyethylene Catalysts
History
Phosphinimide ligands have shown recent promise in the area of ethylene polymerization. Since 1955 this field has been dominated by metallocene based catalysts starting with the Ziegler catalyst, followed shortly by the kaminsky catalyst in 1976.[6][7] Initially phosphinimide ligands were suggested for polyethylene synthesis due to the fact they have similar steric and electronic properties to metallocene polyethylene catalysts.[8]
Phosphinimide Ligands Compared to Cyclopentadienyl Ligands in Ethylene Polymerization
In most respects the sterics and electronic properties of phosphinimide ligands are considered analogous to Cylclopentadienyl ligands.[8][9][10] Sterically, similar cone angles are observed between the metal bound phosphinimide ligand t-Bu3PN (angle of 87°) and the cylclopentadienyl (angle of 83°) suggesting they occupy similar area[8] around the metal centre. However, this does not take into account the fact that the bulkyness of the phosphinimide ligand is somewhat removed from the metal by the distance of the nitrogen metal bond. This removal of bulkyness represents one of the strategies to increase polymerization, that is to increase the exposure of the metal centre.[8] The consequence of a less sterically crowded metal centre appears to be increased deactivation of the catalysis.[8][9][10]
In order to determine the efficiency of each ligand on catalysis, experiments must be performed to determine the rate of reactions and quality of product. Experimentally, ethylene catalysis done by phosphinimide ligand complexes showed similar reactivity when compared to those done with classical cyclopentadienyl lingands. A slightly lower reaction rate and poly ethylene molecular weight where obtained using phosphinimide ligands however, a lower PDI was observed. In terms of ethylene polymerization, phosphinimide ligand catalysis are much more recently developed and therefore further development is likely to increase this catalytic rate.
The phosphinimide ligands provide an alternative to metallocene catalysts due to their capability to be easily altered during synthesis. This capability allows for immense variation in the steric and electronic properties of the system. Experimentally, steric modifications of phosphinimide ligands have been shown to affect the rate of catalysis much more than electronic modifications.[8] In general, the freedom of ligand design can allow for increased specificity of the catalytic properties along with increased ability to obtain mechanistic and catalytic studies of PE synthesis. Also, with an introduction of a phosphorus into the complex, there is one more tool to probe catalytic intermediates in 31P nmr analysis.
General Catalysis Scheme
Activation
The catalyst must be activated in order for polymerization to occur. MAO is a common cocatalyst (activator) for PE polymerization. The discrete structure of MAO is unknown along with its mechanism of activation; however, a general activation scheme has been proposed[6][7][11] primarily to represent the inactive and active species.
Another scheme uses a strong organo-lewis acid such as B(C6F5)3 as a cocatalyst to activate polymerization through methyl abstraction[7][10] thereby producing a highly reactive zwitterion form.[10] Different methods of activation have been shown to produce different ethylene polymerization activity[8][10] making it a very important concept to analyze. In general, it is experimentally observed that phosphinimide catalysts show lower activity when activated by MAO.[10]
Mechanism of Polymerization
The phosphinimide catalyst is thought to be homogeneous and single sited. It therefore produces reactivity comparable to metallocene catalysts[8][10] which are also believed to be homogeneous, single sited catalysts. The catalytic process is assumed to proceed in much of the same way as metallocene based catalysts, as the chemistry is thought to take place primarily with the metal centre and not through the bulky ligands.[9][11]
Deactivation
Catalytic degradation most commonly occurs from cationic dimerization, lewis acid (often with AlMe3 cocatalyst (MAO)) activation of a C-H bond or by excess boron (from the cocatalysts B(C6F5)3) reacting with the active zwitterion.[8][10] Catalytic degradation has been shown to decrease with bulkier phosphinimide ligands[8][10] as they can prevent undesirable interactions.
References
- 1 2 3 4 5 6 Dehnicke, Kurt; Krieger, M.; Massa, W. (1999). "Phosphoraneiminato complexes of transition metals". Coordination Chemistry Reviews. 182: 19–65. doi:10.1016/S0010-8545(98)00191-X.
- 1 2 3 Stephan, Douglas (2006). "Sterically Demanding Phosphinimides: Ligands for Unique Main Group and Transition Metal Chemistry". Advances in Organometallic Chemistry. 54: 267–291. doi:10.1016/S0065-3055(05)54006-1.
- 1 2 3 4 5 6 7 8 Stephan, Douglas W. (2005). "The Road to Early-Transition-Metal Phosphinimide Olefin Polymerization Catalysts". Organometallics. 24 (11): 2548–2560. doi:10.1021/om050096b.
- 1 2 3 4 5 6 7 8 9 10 Dehnicke, Kurt; Weller, F. (1997). "Phosphorane iminato complexes of main group elements". Coordination Chemistry Reviews. 158 (1): 103–169. doi:10.1016/S0010-8545(96)01257-X.
- ↑ Courtenay, Silke; Wei, P.; Stephan, D. (2003). "The syntheses and structures of lithium phosphinimide and phosphinimine complexes". Can. J. Chem. 81: 1471–1476. doi:10.1139/V03-162.
- 1 2 Reddy,S; Sivaram, S. Homogeneous Metallocene-Methylaluminoxane Catalyst Systems for Ethylene Polymerization. Prog. Polym. Sci., 1995, 20, pp 309-367
- 1 2 3 Yue, N.Chemistry of Group (IV) Phosphinimide Complexes. University of Windsor Ontario, Library and Archives Canada, 2000. ISBN 0-494-04962-6
- 1 2 3 4 5 6 7 8 9 10 Stephan,D. ; Stewart.J. et al. An Approach to Catalyst Design: Cyclopentadienyl-Titanium Phosphinimide Complexes in Ethylene Polymerization. Organometallics, 2003, 22, pp 1937-47 DOI: 10.1021/om020954t
- 1 2 3 Voth,P. ; Fraser.C. et al. Functionalizing Titanium-Phosphinimide Complexes. Organometallics, 2006, 25, pp 4779-86 DOI: 10.1021/om060565p
- 1 2 3 4 5 6 7 8 9 Stephan,D. Alcan Award Lecture from Academic Research to Industrial Applications and back Again. Can. J. Chem., 2002, 80, pp 125-32 DOI: 10.1139/V01-203
- 1 2 Hunt, P. Titanium Phosphinimide Polymerization Catalysis. Hunt Research Group, 2006. http://www.huntresearchgroup.org.uk/teaching/teaching_comp_chem_year4/Case_3_TiCat.pdf