1q21.1 deletion syndrome
| 1q21.1 deletion syndrome | |
|---|---|
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q93.5 |
| OMIM | 612474 |
| MedlinePlus | 1q211-microdeletion |
1q21.1 deletion syndrome or 1q21.1 (recurrent) microdeletion is a rare aberration of chromosome 1.
A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.
In 1q21.1, the '1' stands for chromosome 1, the 'q' stands for the long arm of the chromosome and '21.1' stands for the part of the long arm in which the deletion is situated.
The syndrome is a form of the 1q21.1 copy number variations and it is a deletion in the distal area of the 1q21.1 part. The CNV leads to a very variable phenotype and the manifestations in individuals are quite variable. Some people who have the syndrome can function in a normal way, while others have symptoms of mental retardation and various physical anomalies.
1q21.1 microdeletion is a very rare chromosomal condition. Only 46 individuals with this deletion have been reported in medical literature as of August 2011.[1]
Cause of the syndrome
Meiosis is the process of dividing cells in humans. In meiosis, the chromosome pairs split and a representative of each pair goes to one daughter cell. In this way the number of chromosomes will be halved in each cell, while all the parts on the chromosome (genes) remain, after being randomized. Which information of the parent cell ends up in the daughter cell is purely decided by chance. Besides this random process, there is a second random process. In this second random process the DNA will be scrambled in a way that pieces are omitted (deletion), added (duplication), moved from one place to another (translocation) and inverted (inversion). This is a common process, which leads to about 0.4% variation in the DNA.
A problem of the second random process is that genetic mistakes can occur. Because of the deletion and duplication process, the chromosomes that come together in a new cell may be shorter or longer. The result of this spontaneous change in the structure of DNA is a so-called copy number variation. Due to the copy number variation chromosomes of different sizes can be combined in a new cell. If this occurs around conception, the result will be a first cell of a human with a genetic variation. This can be either positive or negative. In positive cases this new human will be capable of a special skill that is assessed positively, for example, in sports or science. In negative cases, you have to deal with a syndrome or a severe disability, as in this case the 1q21.1 deletion syndrome.
Based on the meiotic process, the syndrome may occur in two ways.
- a spontaneous deviation (a 'de novo' situation): two chromosomes come together of which one has a copy number variation as a result of the meiosis process.
- a parent is unknowingly the carrier of a chromosome with a copy number variation and passes it at conception to the child, with different consequences for the child.
Due to this genetic misprint, the embryo may experience problems in the development during the first months of pregnancy. Approximately 20 to 40 days after fertilization, something goes wrong in the construction of the body parts and brain, which leads to a chain reaction.[2]
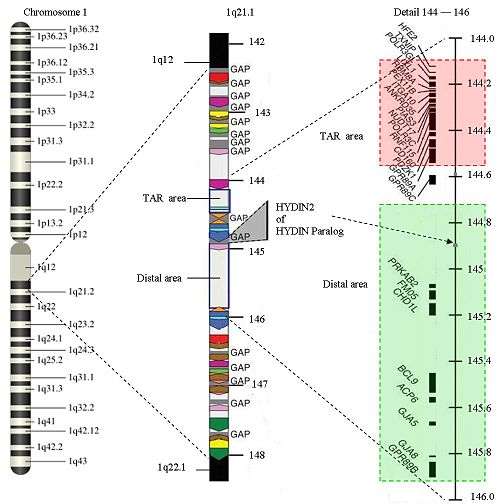
The structure of 1q21.1
The structure of 1q21.1 is complex. The area has a size of approximately 6 Megabase (Mb) (from 141.5 Mb to 147.9 Mb). Within 1q21.1 there are two areas where the CNVs can be found: the proximal area or TAR area (144.1 to 144.5) and the distal area (144.7 to 145.9). The 1q21.1 deletion syndrome will commonly be found in the distal area, but an overlap with the TAR-area is possible. 1q21.1 has multiple repetitions of the same structure (areas with the same color in the picture have equal structures) Only 25% of the structure is not duplicated. There are several gaps in the sequence. There is no further information available about the DNA-sequence in those areas up till now. The gaps represent approximately 700 Kilobase. New genes are expected in the gaps. Because the gaps are still a topic of research, it is hard to find the exact start and end markers of a deletion. The area of 1q21.1 is one of the most difficult parts of the human genome to map.
Because of the repetitions in 1q21.1, there is a larger chance of an unequal crossing-over during meiosis. CNVs occur due to non-allelic homologous recombination mediated by low copy repeats (sequentially similar regions).
Typing
A common deletion is restricted to the distal area. This is a Class I-deletion.
In some cases the deletion is so large that the proximal area is involved as well, the so-called Class II-deletion. There are some complex cases in which both the proximal area and the distal area are affected, while the area in between is normal. There are also some a-typical variants.
Symptoms
Recognised symptoms are:
- Only one set of genes on the two chromosomes function (Haploinsufficiency)
- Thrombocytopenia-absent radius (TAR syndrome), in case of a class II-deletion
- Neurological-psychiatric problems: Autism; schizophrenia;[3][4] epilepsy; learning problems; cognitive disabilities — mild to moderate; developmental delay — mild to moderate (milestones like sitting, standing and walking; come at a later period in childhood); children show an ataxic gait and fall down a lot
- Dysmorphism: Slightly unusual facial appearance; disturbed growth; skeletal malformations; small head (microcephaly); prominent forehead; bulbous nose; deep-set eyes; broad thumbs; broad toes; squint; very flexible joints; clavicular pseudoarthrosis[5] (the collarbone doesn't develop normally) (Class II-deletion); An extra transverse crease of the fifth finger[5] (Class II-deletion)); Problems with the development of the vagina (Müllerian aplasia)
- Eyes: Cataracts
- Heart abnormalities and cardiovascular anomalies (30% of the cases): Anomalous origin of the coronary artery[5] (Class II-deletion)
- Kidneys: Missing kidney or floating kidneys
- Cancer: Neuroblastoma[6]
- Sleep disturbances
It is not clear whether the list of symptoms is complete. Very little information is known about the syndrome. The syndrome can have completely different effects on members of the same family.
A common deletion is between 1.0–1.9Mb. Mefford states that the standard for a deletion is 1.35Mb.[7] The largest deletion seen on a living human is over 5 Mb.
Related genes
Genes related to 1q21.1 deletion in the distal area are PDE4DIP, HYDIN2, PRKAB2, PDIA3P, FMO5, CHD1L, BCL9, ACP6, GJA5, GJA8, NBPF10, GPR89B, GPR89C, PDZK1P1 and NBPF11.
Diagnostics and prevalence
A 'de novo'-situation appears in about 75% of the cases. In 25% of the cases, one of the parents is carrier of the syndrome, without any effect on the parent. Sometimes adults have mild problems with the syndrome. To find out whether either of the parents carries the syndrome, both parents have to be tested. In several cases, the syndrome was identified with the child, because of an autism disorder or another problem, and later it appeared that the parent was affected as well. In families where both parents have tested negative for the syndrome, chances of a second child with the syndrome are extremely low. If the syndrome was found in the family, chances of a second child with the syndrome are 50%, because the syndrome is autosomal dominant. The effect of the syndrome on the child cannot be predicted.
As of October 2012, Unique, an international rare chromosome disorder group and registry, has 64 genetically-confirmed cases of this deletion worldwide.[8]
The Syndrome can be detected with fluorescence in situ hybridization.
For parents with a child with the syndrome, it is advisable to consult a physician before another pregnancy.
Management
Treatment of cause: Due to the genetic cause, no treatment of the cause is possible.
Treatment of manifestations: routine treatment of ophthalmologic, cardiac, and neurologic findings; speech, occupational, and physical therapies as appropriate; specialized learning programs to meet individual needs; antiepileptic drugs or antipsychotic medications as needed.
Surveillance: routine pediatric care; routine developmental assessments; monitoring of specific identified medical issues.
Research
On several locations in the world people are studying on the subject of 1q21.1 deletion syndrome. The syndrome was identified for the first time with people with heart abnormalities. The syndrome has later been found with patients with autism and schizophrenia. Research is done on patients with a symptom of the syndrome, to find more patients with the syndrome.
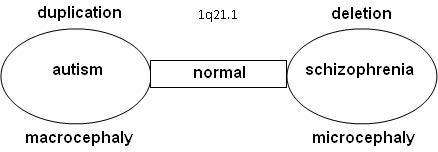
There may be a relation between autism and schizophrenia. Literature shows that nine locations have been found on the DNA where the syndromes related to autism or schizophrenia can be found, the so-called "hotspots": 1q21.1, 3q29, 15q13.3, 16p11.2, 16p13.1, 16q21, 17p12, 21q11.2 and 21q13.3. With a number of hotspots both autism and schizophrenia were observed at that location. In other cases, either autism or schizophrenia has been seen.
Statistical research showed that schizophrenia is more common in combination with 1q21.1 deletion syndrome. On the other side, autism is significantly more common with 1q21.1 duplication syndrome. Further research confirmed that the odds on a relation between schizophrenia and deletions at 1q21.1, 3q29, 15q13.3, 22q11.21 en Neurexin 1 (NRXN1) and duplications at 16p11.2 are at 7.5% or higher.[9][10]
Common variations in the BCL9 gene, which is in the distal area, confer risk of schizophrenia and may also be associated with bipolar disorder and major depressive disorder.[11]
Research is done on 10–12 genes on 1q21.1 that produce DUF1220-locations. DUF1220 is an unknown protein, which is active in the neurons of the brain near the neocortex. Based on research on apes and other mammals, it is assumed that DUF1220 is related to cognitive development (man: 212 locations; chimpanzee: 37 locations; monkey: 30 locations; mouse: 1 location). It appears that the DUF1220-locations on 1q21.1 are in areas that are related to the size and the development of the brain. The aspect of the size and development of the brain is related to autism (macrocephaly) and schizophrenia (microcephaly). It has been proposed that a deletion or duplication of a gene that produces DUF1220-areas might cause growth and development disorders in the brain [12]
Another relation between macrocephaly with duplications and microcephaly with deletions has been seen in research on the HYDIN Paralog or HYDIN2. This part of 1q21.1 is involved in the development of the brain. It is assumed to be a dosage-sensitive gene. When this gene is not available in the 1q21.1 area, it leads to microcephaly. HYDIN2 is a recent duplication (found only in humans) of the HYDIN gene found on 16q22.2.[13] Research on the genes CHD1L and PRKAB2 within lymphoblast cells [14] lead to the conclusion that anomalies appear with the 1q21.1-deletionsyndrome:
- CHD1L is an enzyme which is involved in untangling the chromatides and the DNA repair system. With 1q21.1 deletion syndrome a disturbance occurs, which leads to increased DNA breaks. The role of CHD1L is similar to that of helicase with the Werner syndrome
- PRKAB2 is involved in maintaining the energy level of cells. With 1q21.1-deletion syndrome this function was attenuated.
GJA5 has been identified as the gene that is responsible for the phenotypes observed with congenital heart diseases on the 1q21.1 location. In case of a duplication of GJA5 tetralogy of Fallot is more common. In case of a deletion other congenital heart diseases than tetralogy of Fallot are more common.[15]
References
- ↑ "Overview: 1q21.1 microdeletion syndrome". Genetic and Rare Diseases Information Center (GARD). Office of Rare Diseases Research • U.S. National Institutes of Health. 8 August 2011. Retrieved 9 September 2013.
- ↑ A. Ploeger; 'Towards an integration of evolutionary psychology and developmental science: New insights from evolutionary developmental biology'
- ↑ Stefansson H, Rujescu D, Cichon S, et al. (September 2008). "Large recurrent microdeletions associated with schizophrenia". Nature. 455 (7210): 232–6. doi:10.1038/nature07229. PMC 2687075
 . PMID 18668039.
. PMID 18668039. - ↑ The International Schizophrenia Consortium (September 2008). "Rare chromosomal deletions and duplications increase risk of schizophrenia". Nature. 455 (7210): 237–41. doi:10.1038/nature07239. PMC 3912847. PMID 18668038.
- 1 2 3 Velinov M, Dolzhanskaya N (2010). "Clavicular pseudoarthrosis, anomalous coronary artery and extra crease of the fifth finger-previously unreported features in individuals with class II 1q21.1 microdeletions". Eur J Med Genet. 53 (4): 213–6. doi:10.1016/j.ejmg.2010.05.005. PMID 20573555.
- ↑ Diskin SJ, Hou C, Glessner JT, et al. "Copy number variation at 1q21.1 associated with neuroblastoma". Nature. 459 (7249): 987–991. doi:10.1038/nature08035. PMC 2755253. PMID 19536264.
- ↑ Mefford HC, Sharp AJ, Baker C, et al. (October 2008). "Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes". N. Engl. J. Med. 359 (16): 1685–99. doi:10.1056/NEJMoa0805384. PMC 2703742. PMID 18784092.
- ↑ "Chromosome 1 - 1q21.1 microdeletion" (pdf). rarechromo.org. Unique. 2012. Retrieved 9 September 2013.
- ↑ Levinson DF, Duan J, Oh S, et al. (March 2011). "Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications". Am J Psychiatry. 168 (3): 302–16. doi:10.1176/appi.ajp.2010.10060876. PMID 21285140.
- ↑ Ikeda M, Aleksic B, Kirov G, et al. (February 2010). "Copy number variation in schizophrenia in the Japanese population". Biol. Psychiatry. 67 (3): 283–6. doi:10.1016/j.biopsych.2009.08.034. PMID 19880096.
- ↑ Li J, Zhou G, Ji W, et al. (March 2011). "Common variants in the BCL9 gene conferring risk of schizophrenia". Arch. Gen. Psychiatry. 68 (3): 232–40. doi:10.1001/archgenpsychiatry.2011.1. PMID 21383261.
- ↑ e.g.: Dumas L, Sikela JM (2009). "DUF1220 domains, cognitive disease, and human brain evolution". Cold Spring Harb. Symp. Quant. Biol. 74: 375–82. doi:10.1101/sqb.2009.74.025. PMC 2902282. PMID 19850849.
- ↑ Doggett NA, Xie G, Meincke LJ, et al. (Dec 2006). "A 360-kb interchromosomal duplication of the human HYDIN locus.". Genomics. 88 (6): 762–71. doi:10.1016/j.ygeno.2006.07.012.
- ↑ Understanding the impact of 1q21.1 Copy Number Variant; C. Harvard et al; Orphanet Journal of Rare Diseases 2011, 6:54; doi:10.1186/1750-1172-6-54
- ↑ Soemedi, R.; et al. (2011). "DPhenotype-Specific Effect of Chromosome 1q21.1 Rearrangements and GJA5 Duplications in 2436 Congenital Heart Disease Patients and 6760 Controls". Hum. Mol. Genet. 21: 1513–1520. doi:10.1093/hmg/ddr589.
Further reading
- Genetics of Mental Retardation, Karger. Knight S. (ed) Chapter One: 'A Parent's' Perspective' contains description and photos of female with 1q21.1 microdeletion
- Brunet A, Armengol L, Heine D, et al. (2009). "BAC array CGH in patients with Velocardiofacial syndrome-like features reveals genomic aberrations on chromosome region 1q21.1". BMC Med. Genet. 10: 144. doi:10.1186/1471-2350-10-144. PMC 2805625. PMID 20030804.
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, et al. (2008). "Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities.". Nat Genet. 40 (12): 1466–71. doi:10.1038/ng.279. PMC 2680128. PMID 19029900.
- Crespi B, Stead P, Elliot M (2010). "Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia". Proc Natl Acad Sci U S A. 107 (Suppl 1): 1736–41. doi:10.1073/pnas.0906080106. PMC 2868282. PMID 19955444.
External links
- DECIPHER database entry for 1q21.1 deletion syndrome
- 1q21.1 deletion, GeneReviews NCBI Bookshelf
- Orpha.net
- Blogs on children with 1q21.1 deletion syndrome Chrissy, Laura and Niko