Fibroblast growth factor receptor 1
| View/Edit Human | View/Edit Mouse |
Fibroblast growth factor receptor 1 (FGFR1), also known as basic fibroblast growth factor receptor 1, fms-related tyrosine kinase-2 / Pfeiffer syndrome, and CD331, is a receptor tyrosine kinase whose ligands are specific members of the fibroblast growth factor family. FGFR1 has been shown to be associated with Pfeiffer syndrome.[4]
Function
The protein encoded by this gene is a member of the fibroblast growth factor receptor (FGFR) family, where amino acid sequence is highly conserved between members and throughout evolution. FGFR family members differ from one another in their ligand affinities and tissue distribution. A full-length representative protein consists of an extracellular region, composed of three immunoglobulin-like domains, a single hydrophobic membrane-spanning segment and a cytoplasmic tyrosine kinase domain. The extracellular portion of the protein interacts with fibroblast growth factors, setting in motion a cascade of downstream signals, ultimately influencing mitogenesis and differentiation. This particular family member binds both acidic and basic fibroblast growth factors and is involved in limb induction.
Clinical significance
Mutations in this gene have been associated with Pfeiffer syndrome, Jackson–Weiss syndrome, Antley-Bixler syndrome, Trigonocephaly, osteoglophonic dysplasia, squamous cell lung cancer (14) and autosomal dominant Kallmann syndrome. There is also strong evidence from sequencing studies of candidate genes involved in clefting that mutations in the FGFR1 gene may be associated in the pathogenesis of cleft lip and/or palate.[5] A few DNA sequence variants, including one nonsense mutation, have been reported in isolated or non-syndromic cleft lip and palate.[6] Both cleft lip with or without a cleft palate and cleft palate only features have been seen in families with a FGFRI mutation.[5] Cleft palate is a relatively common feature of Kallman syndrome as well.[5][7][8] Somatic chromosomal aberrations involving this gene are associated with stem cell myeloproliferative disorder and stem cell leukemia lymphoma syndrome. Alternatively spliced variants which encode different protein isoforms have been described; however, not all variants have been fully characterized.[9] Mutations in this gene have been associated to Hartsfield syndrome .[10]
Cancer
Somatic mutations of FGFR1 occurs in several diseases including breast and lung cancers. FGFR1 has an amplification of 9%–15% in breast cancer.[11] Its amplified expression is generally associated with poor prognosis and relapse. In ER+ breast cancer, its amplification is associated with aggressive metastasis. It is also associated as a cancer subtype as amplification of it and ERBB2 is mutually exclusive. Apart from gene amplification, a somatic mutation called K566R has also been associated with basal-like triple negative breast cancer.
Amplification of FGFR1 is also a feature in 20% of lung cancer patients. FGFR1 amplification was a single largest prognostic factor in a cohort of SCLC patients[12] and associated with smoking. It has also been detected amplified in NSCLC cancer patients. Chromosomal translocations are an important factor in FGFR1's involvement in cancer. FN1-FGFR1 fusion is frequent in phosphaturic mesenchymal tumours.[13][14] FGFR1 fusions have been detected with BCR and ZFN198, indicating interactions with conventional oncogenic pathways. There fusion proteins can transform cells and induce SCLL cancer in mice. Multiple myelomas also harbor a t(4;14) translocation of the FGFR1 gene. The translocation brings FGFR in proximity to the IgH enchancer region leading to overexpression. In prostate cancer, expression of FGFR1 leads to an induced EMT of cancer cells.[15] This mechanism probably involves interaction with stromal cancer associated fibroblasts leading to dynamics that stimulates NFkB and as a result cell invasiveness.
The mechanism of FGFR1 action on cancer is largely based on abetting cancer progression and metastasis.[16] Fibroblast growth factors trigger the autophosphorylation of the receptors at important tyrosine residues within the activation loop of the tyrosine kinase domain. This autophosphorylation results in change of the structural conformation of the tyrosine kinase domain from an inactive form to an active one [17] This activated tyrosine kinase domain phosphorylates tyrosine residues at other sites in FGFR1 along with FGFR1-bound adaptor molecules in a stepwise manner. This phosphorylation of the tyrosine residue at the C-terminal region of receptor allows for the binding site of phospholipase Cγ in recruiting and activating PLCγ for the catalysis and transformation of phosphatidylinositol diphosphate (PIP2) to diacylglycerol (DAG) and inositol triphosphate (IP3). FGF signals are downstream effected into the RAS-MAPK or PI3K-AKT signaling cascades.[18] SRC tyrosine kinase are also activated by FGF signals.[19] FGFR1 receives fibroblast binding proteins and concurrently activates both the Ras-MAPK/ERK and the PI3K pathway. Both these pathways are fundamentally important to cancer cells as they provide signals to proliferate and survive respectively. As the control of these pathways is tuned by the presence of the FGF receptor, ordinarily it would not be an issue in epithelial cells. However cancer cells that undergo EMT take up a more messenchymal state.[20] In these cases the FGF receptors are upregulated leading to a greater tendency to move to the messenchymal state and increased aggressiveness as well as motility.
FGFR1 activity also displays focal dependence in terms of ligand signaling. Basal FGFR1 signaling was lower in FGFR1 inhibitor sensitive cell lines such as DMS114. However, on addition of FGFs, the p-ERK pathway was highly stimulated.[21] Under steady state conditions the cell lines exhibited high levels of FGF secretion and under serum starvation conditions the cells still produced above average levels of FGF. This indicates that even under nutrition deficient conditions, FGF secretion and presence of FGFRs on cell surface allow cells to sustain growth. Interestingly, pERK could be activated by FGF1 but FGFR levels were unaffected by the presence of FGF inhibitor. This results to the conclusion that a certain baseline level of FGF activity possibly through autocrine or paracrine signaling sustains the display of FGF receptors even in conditions that normally would not favor growth. Its this counterintuitive presence of receptor that allows cancer cells to grow in a non-growth environment.[22]
FGFR inhibitors
Recent advances in our knowledge of the FGFR1 system has resulted in attempts to discover therapeutic touch points in the pathway for use in drug development. FGFR-targeted drugs exert direct as well as indirect anticancer effects, because FGFRs on cancer cells and endothelial cells are involved in tumorigenesis and vasculogenesis, respectively.[17] FGFR therapeutics are active as FGF affects numerous morphologies of cancers such as invasiveness, stemness and cellular survival. Primary among such drugs are antagonists. Small molecules that fit between the ATP binding pockets of the tyrosine kinase domains of the receptors. For FGFR1 numerous such small molecules are already approved for targeting of the structure of TKI ATP pocket. These include Dovitinib and Brivanib. The table below provides the IC50 (Nanomolar) of Small-Molecule Compounds Targeting FGFRs.[17]
| PD173074 | TKI258 Dovitinib | Ki23057 | E7080 | Brivanib | BIBF1120 | AP24534 Ponatinib | MK-2461 | E-3810 | AZD4547 |
|---|---|---|---|---|---|---|---|---|---|
| 26 | 8 | NA | 46 | 148 | 69 | 2.2 | 65 | 18 | 0.2 |
FGFR1 aberration in breast and lung cancer as a result of genetic overamplification is effectively targeted using Dovitinib and Povatinib respectively.[23] Drug resistance is a highly relevant topic in the field of drug development for FGFR targets. FGFR inhibitors allow for the increase of tumor sensitivity to regular anticancer drug such as paclitaxel, and etoposide in human cancer cells and thereby enhancing antiapoptotic potential based on aberrant FGFR activation.[17] Moreover, FGF signaling inhibition dramatically reduces revascularization, hitting upon one of the hallmarks of cancers, angiogenesis, and reduces tumor burden in human tumors that depend on autocrine FGF signaling based on FGF2 upregulation following the common VEGFR-2 therapy for breast cancer. In such a way, FGFR1 can act synergistically with therapies to cut off cancer clonal resurgence by eliminating potential pathways of future relapse.
In addition, FGFR inhibitors are predicted to be effective on relapsed tumors because of the clonal evolution of an FGFR-activated minor subpopulation after therapy targeted to EGFRs or VEGFRs. Because there are multiple mechanisms of action for FGFR inhibitors to overcome drug resistance in human cancer, FGFR-targeted therapy is a promising strategy for the treatment of refractory cancer.
AZD4547 has undergone a phase II clinical trial (re gastric cancer) and reported some results.[24]
Lucitanib is an inhibitor of FGFR1 and FGFR2 and has undergone clinical trials for advanced solid tumors.[25]
Dovitinib (TKI258), an inhibitor of FGFR1, FGFR2, and FGFR3, has had a clinical trial on FGFR-amplified breast cancers.[26]
Interaction with PLCγ
Receptor tyrosine kinase (RTK) signaling plays an important role in various signaling pathways in human biology. The phosphorylation of tyrosines at the juxtamembrane region, kinase insert, or C-terminal tail of RTKs creates a docking site for the binding of Src Homology 2 (SH2) domain in RTK signaling pathways. SH2 domain recognizes the phosphorylated tyrosine residues (pTyr) on its substrate to activate a specific intracellular signaling pathway. Previous studies have also shown that RTKs utilize their unique autophosphorylation properties to recruit target proteins upon ligand stimulation.
The phosphorylation of a highly conserved tyrosine residue (Y766) at the C-terminal tail of Fibroblast growth factor 1 (FGFR1) creates a binding site to recruit Phospholipase C gamma (PLCγ), a tandem nSH2- cSH2 containing domain, to facilitate phosphorylation of PLCγ. Phosphorylated PLCγ relieves its autoinhibition, facilitates the enzymatic activity to hydrolyze phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and second messenger diacyglycerol (DAG). These molecules then serves as a second messenger for other signaling pathways.
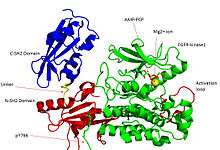
Recent publication on the 2.5 Å crystal structure of PLCγ in complex with FGFR1 kinase (PDB: 3GQI) provides new insights in understanding the molecular mechanism of substrate recruitment by SH2 domain to its substrate. Figure 1 shows the PLCγ-FGFR1 kinase complex with the c-SH2 domain colored in red, n-SH2 domain colored in blue, and the interdomain linker is colored in yellow. The structure contains typical SH2 domain, with two α-helices and three antiparallel β-strands on each SH2 domain. In this complex, the phosphorylated tyrosine (pY766) on the C-terminal tail of FGFR1 kinase binds preferentially to the nSH2 domain of PLCγ. The phosphorylation of tyrosine residue 766 on FGFR1 kinase forms hydrogen bonds with the n-SH2 to stabilize the complex. Hydrogen bonds in the binding pocket help to stabilize the PLCγ-FGFR1 kinase complex. The water molecule as shown mediates the interaction of asparagine 647 (N647) and aspartate 768 (D768) to further increase the binding affinity of the n-SH2 and FGFR1 kinase complex. (Figure 2).
The phosphorylation of tyrosine 653 and tyrosine 654 in the active kinase conformation causes a large conformation change in the activation segment of FGFR1 kinase. Threonine 658 is moved by 24Å from the inactive form (Figure 3.) to the activated form of FGFR1 kinase (Figure 4.). The movement causes the closed conformation in the inactive form to open to enable substrate binding. It also allows the open conformation to coordinate Mg2+ with AMP-PCP (analog of ATP). In addition, pY653 and pY654 in the active form helps to maintain the open conformation of the SH2 and FGFR1 kinase complex. However, the mechanism of which the phosphorylation at Y653 and Y654 helps to recruit SH2 domain to its C-terminal tail upon phosphorylation of Y766 remains elusive. Figure 5 shows the overlay structure of active and inactive forms of FGFR1 kinase. Figure 6 shows the dots and contacts on phosphorylated tyrosine residues 653 and 654. Green dots show highly favorable contacts between pY653 and pY654 with surrounding residues. Red spikes show unfavorable contacts in the activation segment. The figure is generated through Molprobity extension on Pymol.
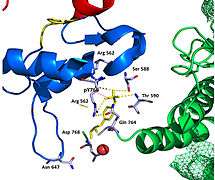 Figure 2. Hydrogen bonds at pY766
Figure 2. Hydrogen bonds at pY766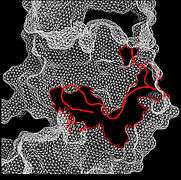 Figure 3. Closed conformation in Inactive FRFR1 kinase
Figure 3. Closed conformation in Inactive FRFR1 kinase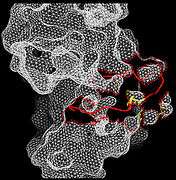 Figure 4. Open conformation in active FRFR1 kinase
Figure 4. Open conformation in active FRFR1 kinase Figure 5. Overlay Structures of Active and Inactive Forms of FGFR1 kinase
Figure 5. Overlay Structures of Active and Inactive Forms of FGFR1 kinase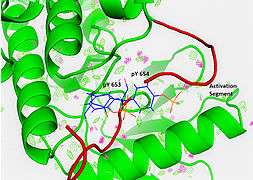 Figure 6. Dots and contacts on pY653 & pY654
Figure 6. Dots and contacts on pY653 & pY654 Figure 7. The β-factor of the PLC-FGFR1 kinase complex
Figure 7. The β-factor of the PLC-FGFR1 kinase complex
N-SH2 binding site interface
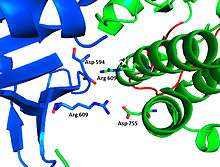
The FGFR1 kinase is bound to the N-SH2 domain primarily through charged amino acids. Arginine residue (R609) on the N-SH2 domain forms a salt bridge to aspartate 755 (D755) on the FGFR1 domain. The acid base pair located in the middle of the interface are nearly parallel to each other, indicating a highly favorable interaction. N-SH2 domain makes an additional polar contact through water-mediated interaction that takes place between the N-SH2 domain and FGFR1 kinase. Interestingly, the arginine residue 609 (R609) on the FGFR1 kinase also forms a salt bridge to the aspartate residue (D594) on the N-SH2 domain. The acid-base pair interacts with each other carry out redox reaction to stabilize the complex (Figure 7). Previous studies have done to elucidate the binding affinity of the n-SH2 domain with the FGFR1 kinase complex by mutating these amino acids phenyalanine or valine. The results from isothermal titration calorimetry (ITC) indicated that the binding affinity of the complex decreased by 3 to 6-fold, without affecting the phosphorylation of the tyrosine residues. [27]
Interactions
Fibroblast growth factor receptor 1 has been shown to interact with:
See also
References
- ↑ "Drugs that physically interact with Fibroblast growth factor receptor 1 view/edit references on wikidata".
- ↑ "Human PubMed Reference:".
- ↑ "Mouse PubMed Reference:".
- ↑ Itoh N, Terachi T, Ohta M, Seo MK (Jun 1990). "The complete amino acid sequence of the shorter form of human basic fibroblast growth factor receptor deduced from its cDNA". Biochemical and Biophysical Research Communications. 169 (2): 680–5. doi:10.1016/0006-291X(90)90384-Y. PMID 2162671.
- 1 2 3 Dixon MJ, Marazita ML, Beaty TH, Murray JC (Mar 2011). "Cleft lip and palate: understanding genetic and environmental influences". Nature Reviews Genetics. 12 (3): 167–78. doi:10.1038/nrg2933. PMC 3086810
 . PMID 21331089.
. PMID 21331089. - ↑ Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dodé C, Mohammadi M, Marazita ML, Murray JC (Mar 2007). "Impaired FGF signaling contributes to cleft lip and palate". Proceedings of the National Academy of Sciences of the United States of America. 104 (11): 4512–7. Bibcode:2007PNAS..104.4512R. doi:10.1073/pnas.0607956104. JSTOR 25426864. PMC 1810508. PMID 17360555.
- ↑ Kim HG, Herrick SR, Lemyre E, Kishikawa S, Salisz JA, Seminara S, MacDonald ME, Bruns GA, Morton CC, Quade BJ, Gusella JF (Aug 2005). "Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FGFR1". Journal of Medical Genetics. 42 (8): 666–72. doi:10.1136/jmg.2004.026989. PMC 1736121. PMID 16061567.
- ↑ Dodé C, Fouveaut C, Mortier G, Janssens S, Bertherat J, Mahoudeau J, et al. (Jan 2007). "Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis". Human Mutation. 28 (1): 97–8. doi:10.1002/humu.9470. PMID 17154279.
- ↑ EntrezGene 2260
- ↑ Dhamija R, Kirmani S, Wang X, Ferber MJ, Wieben ED, Lazaridis KN, Babovic-Vuksanovic D (Sep 2014). "Novel de novo heterozygous FGFR1 mutation in two siblings with Hartsfield syndrome: a case of gonadal mosaicism". American Journal of Medical Genetics Part A. 164A (9): 2356–9. doi:10.1002/ajmg.a.36621. PMID 24888332.
- ↑ https://pct.mdanderson.org/genes/fgfr1/show[]
- ↑ Kim HR, Kim DJ, Kang DR, Lee JG, Lim SM, Lee CY, Rha SY, Bae MK, Lee YJ, Kim SH, Ha SJ, Soo RA, Chung KY, Kim JH, Lee JH, Shim HS, Cho BC (Feb 2013). "Fibroblast growth factor receptor 1 gene amplification is associated with poor survival and cigarette smoking dosage in patients with resected squamous cell lung cancer". Journal of Clinical Oncology. 31 (6): 731–7. doi:10.1200/JCO.2012.43.8622. PMID 23182986.
- ↑ Wasserman JK, Purgina B, Lai CK, Gravel D, Mahaffey A, Bell D, Chiosea SI (Jan 2016). "Phosphaturic Mesenchymal Tumor Involving the Head and Neck: A Report of Five Cases with FGFR1 Fluorescence In Situ Hybridization Analysis". Head and Neck Pathology. doi:10.1007/s12105-015-0678-1. PMID 26759148.
- ↑ Lee JC, Jeng YM, Su SY, Wu CT, Tsai KS, Lee CH, et al. (Mar 2015). "Identification of a novel FN1-FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour". The Journal of Pathology. 235 (4): 539–45. doi:10.1002/path.4465. PMID 25319834.
- ↑ Wan X, Corn PG, Yang J, Palanisamy N, Starbuck MW, Efstathiou E, et al. (Sep 2014). "Prostate cancer cell-stromal cell crosstalk via FGFR1 mediates antitumor activity of dovitinib in bone metastases". Science Translational Medicine. 6 (252): 252ra122. doi:10.1126/scitranslmed.3009332. PMID 25186177.
- ↑ Yang F, Zhang Y, Ressler SJ, Ittmann MM, Ayala GE, Dang TD, Wang F, Rowley DR (Jun 2013). "FGFR1 is essential for prostate cancer progression and metastasis". Cancer Research. 73 (12): 3716–24. doi:10.1158/0008-5472.CAN-12-3274. PMC 3686853. PMID 23576558.
- 1 2 3 4 Katoh M, Nakagama H (Mar 2014). "FGF receptors: cancer biology and therapeutics". Medicinal Research Reviews. 34 (2): 280–300. doi:10.1002/med.21288. PMID 23696246.
- ↑ "FGFR1 (Fibroblast Growth Factor Receptor 1)". atlasgeneticsoncology.org. Retrieved 30 May 2015.
- ↑ "Src activation plays an important key role in lymphomagenesis induced by FGFR1-fusion kinases". PubMed Central (PMC). 71: 7312–22. 2011. doi:10.1158/0008-5472.CAN-11-1109. PMC 3228896. PMID 21937681.
- ↑ "DEFINE_ME_WA". cell.com. Retrieved 30 May 2015.
- ↑ Malchers F, et al. (Feb 2014). "Cell-autonomous and non-cell-autonomous mechanisms of transformation by amplified FGFR1 in lung cancer.". nih.gov. 4: 246–57. doi:10.1158/2159-8290.CD-13-0323. PMID 24302556.
- ↑ Malchers F, Dietlein F, Schöttle J, Lu X, Nogova L, Albus K, et al. (Feb 2014). "Cell-autonomous and non-cell-autonomous mechanisms of transformation by amplified FGFR1 in lung cancer". Cancer Discovery. 4 (2): 246–57. doi:10.1158/2159-8290.CD-13-0323. PMID 24302556.
- ↑ André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. (2013). "Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer". Clinical Cancer Research : an Official Journal of the American Association for Cancer Research. 19 (13): 3693–702. doi:10.1158/1078-0432.CCR-13-0190. PMID 23658459.
- ↑ A randomized, open-label phase II study of AZD4547 (AZD) versus Paclitaxel (P) in previously treated patients with advanced gastric cancer (AGC) with Fibroblast Growth Factor Receptor 2 (FGFR2) polysomy or gene amplification (amp): SHINE study.
- ↑ Soria; et al. (2014). "Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors.". doi:10.1093/annonc/mdu390. PMID 25193991.
- ↑ Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer.
- ↑ Bae JH, Lew, ED, Yuzawa S, Tome F, Lax I, Schlessinger J (Aug 2009). "The Selectivity of Receptor Tyrosine Kinase Signaling Is Controlled by a Secondary SH2 Domain Binding Site". Cell. 138 (3): 514–24. doi:10.1016/j.cell.2009.05.028. PMC 4764080. PMID 19665973.
- ↑ Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M (Sep 2000). "Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization". Mol. Cell. 6 (3): 743–50. doi:10.1016/s1097-2765(00)00073-3. PMID 11030354.
- ↑ Santos-Ocampo S, Colvin JS, Chellaiah A, Ornitz DM (Jan 1996). "Expression and biological activity of mouse fibroblast growth factor-9". J. Biol. Chem. 271 (3): 1726–31. doi:10.1074/jbc.271.3.1726. PMID 8576175.
- ↑ Yan KS, Kuti M, Yan S, Mujtaba S, Farooq A, Goldfarb MP, Zhou MM (May 2002). "FRS2 PTB domain conformation regulates interactions with divergent neurotrophic receptors". J. Biol. Chem. 277 (19): 17088–94. doi:10.1074/jbc.M107963200. PMID 11877385.
- ↑ Ong SH, Guy GR, Hadari YR, Laks S, Gotoh N, Schlessinger J, Lax I (Feb 2000). "FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors". Mol. Cell. Biol. 20 (3): 979–89. doi:10.1128/mcb.20.3.979-989.2000. PMC 85215. PMID 10629055.
- ↑ Xu H, Lee KW, Goldfarb M (Jul 1998). "Novel recognition motif on fibroblast growth factor receptor mediates direct association and activation of SNT adapter proteins". J. Biol. Chem. 273 (29): 17987–90. doi:10.1074/jbc.273.29.17987. PMID 9660748.
- ↑ Dhalluin C, Yan KS, Plotnikova O, Lee KW, Zeng L, Kuti M, Mujtaba S, Goldfarb MP, Zhou MM (Oct 2000). "Structural basis of SNT PTB domain interactions with distinct neurotrophic receptors". Mol. Cell. 6 (4): 921–9. doi:10.1016/S1097-2765(05)00087-0. PMID 11090629.
- ↑ Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (Dec 2006). "Klotho converts canonical FGF receptor into a specific receptor for FGF23". Nature. 444 (7120): 770–4. Bibcode:2006Natur.444..770U. doi:10.1038/nature05315. PMID 17086194.
- ↑ Reilly JF, Mickey G, Maher PA (Mar 2000). "Association of fibroblast growth factor receptor 1 with the adaptor protein Grb14. Characterization of a new receptor binding partner". J. Biol. Chem. 275 (11): 7771–8. doi:10.1074/jbc.275.11.7771. PMID 10713090.
- ↑ Karlsson T, Songyang Z, Landgren E, Lavergne C, Di Fiore PP, Anafi M, Pawson T, Cantley LC, Claesson-Welsh L, Welsh M (Apr 1995). "Molecular interactions of the Src homology 2 domain protein Shb with phosphotyrosine residues, tyrosine kinase receptors and Src homology 3 domain proteins". Oncogene. 10 (8): 1475–83. PMID 7537362.
Further reading
- Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. (Dec 2010). "Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer". Science Translational Medicine. 2 (62): 62ra93. doi:10.1126/scitranslmed.3001451. PMC 3990281. PMID 21160078.
- Johnson DE, Williams LT (1992). "Structural and Functional Diversity in the FGf Receptor Multigene Family". Advances in Cancer Research. 60. pp. 1–41. doi:10.1016/S0065-230X(08)60821-0. ISBN 978-0-12-006660-5. PMID 8417497.
- Macdonald D, Reiter A, Cross NC (2002). "The 8p11 myeloproliferative syndrome: a distinct clinical entity caused by constitutive activation of FGFR1". Acta Haematologica. 107 (2): 101–7. doi:10.1159/000046639. PMID 11919391.
- Groth C, Lardelli M (2002). "The structure and function of vertebrate fibroblast growth factor receptor 1". The International Journal of Developmental Biology. 46 (4): 393–400. PMID 12141425.
- Wilkie AO (Apr 2005). "Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations". Cytokine & Growth Factor Reviews. 16 (2): 187–203. doi:10.1016/j.cytogfr.2005.03.001. PMID 15863034.
External links
- GeneReviews/NIH/NCBI/UW entry on FGFR-Related Craniosynostosis Syndromes
- GeneReviews/NCBI/NIH/UW entry on Kallmann syndrome
- FGFR1 protein, human at the US National Library of Medicine Medical Subject Headings (MeSH)
- Fibroblast growth factor receptor 1 on the Atlas of Genetics and Oncology
- FGFR1 human gene location in the UCSC Genome Browser.
- FGFR1 human gene details in the UCSC Genome Browser.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||